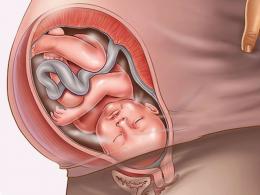
Церебральный амилоидоз. Амилоидоз подтип аа почему неизлечим. Ультразвуковое исследование внутренних органов
Амилоидоз - заболевание, характеризующееся нарушением обмена веществ, в результате чего образуется новое для организма вещество (амилоид), которое откладывается в органах и нарушает их функции.
Амилоид является сложным гликопротеидом, в котором фибриллярные и глобулярные белки тесно связаны с полисахаридами. Фибрилла амилоида состоит из полипептидных белков; кроме фибриллярного белка, в состав амилоида входит и другой белок - так называемый Р-компонент, одинаковый при всех формах амилоида. Предполагают, что Р-компонент является нормальным сывороточным белком, связанным с амилоидными фибриллами.
Амилоидоз может возникать как осложнение каких-либо заболеваний или развиваться как самостоятельный процесс.
В настоящее время в зависимости от этиологии выделяют несколько форм амилоидоза, имеющих свойственный им биохимический состав амилоидных фибрилл.
Первичный (идиопатический) амилоидоз развивается без видимых причин и поражает различные органы (сердце, почки, кишечник, печень, нервную систему). Биохимическая форма первичного амилоидоза - AL-форма, предшественником такого амилоида являются Ig и легкие цепи иммуноглобулинов. По структуре амилоида и характеру поражения внутренних органов к первичному (идиопатическому) амилои-дозу близок амилоидоз при миеломной болезни, который в настоящее время выделяется в отдельную группу.
Наследственный (генетический) амилоидоз проявляется преимущественным поражением почек, сочетанием поражения почек и нервной системы. В нашей стране наследственный амилоидоз обычно связан с периодической болезнью, которая передается по аутосомно-доми-нантному типу. При этом заболевании амилоидоз может быть единственным проявлением. Биохимическая форма наследственного амилоидоза - AF (предшественником амилоида является преальбумин). В случае периодической болезни биохимическая форма - АА (предшественником является белок SAA).
Приобретенный (вторичный) амилоидоз встречается наиболее часто и развивается при ревматоидном артрите, болезни Бехтерева, туберкулезе, хронических нагноениях - остеомиелите, бронхоэкта-тической болезни, хроническом абсцессе легкого, реже - при неспецифическом язвенном колите, псориазе, лимфогранулематозе, сифилисе, опухолях почки, легкого и др. Биохимическая форма вторичного амилоидоза - АА (его сывороточный предшественник - белок SAA, синтезируемый гепатоцитами).
Старческий амилоидоз - результат инволютивных нарушений обмена белка, обнаруживается в головном мозге, поджелудочной железе, сердце. Биохимическая формула - AS (предшественником является преальбумин).
Локальный амилоидоз развивается без видимых причин, его биохимическая формула - АЕ (предшественник неизвестен).
Патогенез. Хорошо известны лишь отдельные звенья патогенеза (схема 22). Из схемы следует, что под влиянием мутации генов, а также воздействия внешних факторов изменяется иммунитет - уменьшается
количество Т-лимфоцитов. Это приводит к снижению контролирующего их воздействия на В-систему лимфоцитов. В результате уменьшается количество В-клеток, несущих нормальные иммуноглобулины, и увеличивается количество В-клеток, синтезирующих предшественников амилоидной фибриллы. Амилоидобласты в повышенном количестве продуцируют фибриллярный белок, что обусловливает синтез амилоида в большом количестве.
Однако вследствие генетического дефекта амилоидокластов, способствующего снижению их ферментативной активности, достаточной резорбции амилоида не происходит. В результате наблюдается усиленное отложение амилоида в тканях и органах [Мухин Н.А., 1981].
При миеломной болезни амилоидоз развивается в результате повышенной продукции плазмоцитами парапротеина, идущего на построение амилоида. Состав амилоида при разных формах амилоидоза различен, что определяется составом белка амилоидных фибрилл.
При поражении миокарда, периферических нервов (наблюдается преимущественно при идиопатической форме амилоидоза) амилоид откладывается вокруг коллагеновых волокон соединительной ткани. Отложение амилоида вокруг ретикулярных волокон наблюдается при поражении
почек, кишечника, печени, надпочечников, поджелудочной железы (при наследственном и вторичном амилоидозе). Однако возможно сочетание периколлагенового и периретикулярного отложения амилоида, что обеспечивает сочетанные поражения различных органов и систем.
При отложении амилоида в тканях уменьшается количество функционирующих элементов, кардиомиоцитов, гепатоцитов, нервных волокон, почечных клубочков, что в последующем приводит к развитию недостаточности органа.
Так, в сердце амилоид откладывается под эндокардом, в строме и сосудах миокарда, а также по ходу вен в эпикарде. Сердце при этом резко увеличивается в размерах, а количество кардиомиоцитов быстро убывает. Все это приводит к снижению сократительной функции миокарда и сердечной недостаточности, а также нарушениям проводимости и ритма сердца. В головном мозге при старческом амилоидозе амилоид находят в так называемых сенильных бляшках коры, сосудах и оболочках. В коже амилоид откладывается в сосочках и стенках сосудов, что приводит к резкой атрофии эпидермиса. В печени амилоид откладывается между звездчатыми ретикулоэндотелиоцитами синусоидных сосудов, в стенках сосудов, протоков, в соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки атрофируются.
В почках амилоид откладывается в мембране клубочковых капилляров и канальцев нефрона, в мезангии, капиллярных петлях и по ходу ар-териол. По мере накопления амилоида большинство нефронов атрофируется, погибает или замещается соединительной тканью - возникает ами-лоидно-сморщенная почка. Этот процесс можно представить в виде следующей схемы:
протеинурия -> нефротический синдром -> почечная недостаточность.
Соответственно этому в клинической картине выделяют три стадии: 1) начальную (протеинурическую); 2) развернутую (нефротическую); 3) терминальную (азотемическую).
Клиническая картина. Проявления амилоидоза разнообразны и определяются: 1) локализацией амилоида в том или ином органе; 2) степенью выраженности отложений амилоида в органе; 3) основным заболеванием, на фоне которого развился амилоид (при вторичной форме амилоидоза).
При диагностике могут возникнуть затруднения, обусловленные тем, что клинические проявления болезни будут заметны лишь при определенном количестве отложившегося амилоида. В связи с этим неизбежен «латентный» период от момента отложения амилоида до появления симптомов нарушения функционирования органа или системы.
Клиническая картина особенно яркая при поражении почек - наиболее частой локализации отложений амилоида.
На I этапе диагностического поиска в начальной стадии практически никакой информации, свидетельствующей о поражении почек ами-лоидозом, получить не удается. Жалобы больных связаны с основным заболеванием (при вторичном амилоидозе).
В анамнезе имеются сведения о наличии того или иного заболевания (туберкулез легких, остеомиелит, ревматоидный артрит и пр.), его течении, проводившейся терапии. Сами по себе эти сведения не позволяют диагностировать амилоидоз почек, но обращают внимание врача на такую возможность.
В развернутой стадии амилоидоза больные предъявляют жалобы, обусловленные развитием нефротического синдрома, на уменьшение количества мочи, отеки различной распространенности и выраженности, а также жалобы на слабость, отсутствие аппетита, снижение работоспособности. Наряду с ними при вторичном амилоидозе остаются жалобы на проявление основного заболевания.
В терминальной стадии жалобы вызваны развивающейся хронической почечной недостаточностью: снижение аппетита, тошнота, рвота (диспепсические расстройства), головные боли, нарушение сна (нарушения нервной системы), кожный зуд.
На II этапе диагностического поиска в ранней стадии могут обнаруживаться только симптомы, характерные для основного заболевания (при вторичном амилоидозе).
В развернутой стадии выявляют: 1) отеки различной локализации и выраженности; при значительной задержке жидкости в организме могут появляться гидроторакс, гидроперикард, преходящий асцит; 2) артериальную гипертензию (встречается у 12 - 20 % больных амилоидо-зом), дилатацию и гипертрофию левого желудочка; 3) увеличение печени и селезенки вследствие отложения в тканях амилоида (печень и селезенка плотные, безболезненные, с заостренным краем); 4) симптомы основного заболевания (при вторичном амилоидозе).
В терминальной стадии симптоматика определяется выраженностью почечной недостаточности: 1) дистрофический синдром (изменения кожи и слизистых оболочек); 2) серозно-суставной синдром (остеоартропатии, вторичная подагра, сухой перикардит, плеврит); 3) артериальная гипертензия.
На III этапе диагностического поиска при амилоидозе получают наиболее значимую для постановки диагноза информацию, которую можно сгруппировать следующим образом: 1) мочевой синдром; 2) нарушения белкового и липидного обмена; 3) обнаружение отложения амилоидных масс.
Мочевой синдром: 1) протеинурия - важнейший симптом амилоидоза, развивается при всех его формах, но наиболее часто при вторичном амилоидозе. Протеинурия обычно бывает значительной, за сутки выделяется 2 -20 г белка, основную часть которого составляют альбумины. В меньших количествах выделяются глобулины, возможно выведение с мочой сывороточного предшественника амилоида (белок SAA). В терминальной стадии протеинурия сохраняется. В моче можно обнаружить а- и особенно у-гликопротеиды.
Соответственно степени протеинурии обнаруживают гиалиновые и реже зернистые цилиндры. Нечасто диагностируется микрогематурия или лейкоцитурия, однако выраженность ее не соответствует степени протеи-нурии (как это наблюдается при гломерулонефритах). Степени нарушений липидного обмена при амилоидозе соответствует липоидурия с наличием двоякопреломляющих кристаллов в осадке мочи.
Нарушения белкового и липидного обмена: 1) гипопротеинемия в сочетании с гипоальбуминемией и гипер-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
Большое значение для диагноза имеет обнаружение амилоидных масс в органах и тканях: в печени (50 % случаев), селезенке (при пункцион-ной биопсии), слизистой оболочке десны и прямой кишки.
В начальной стадии (протеину рической) при биопсии слизистой оболочки десны чаще получают отрицательный, а прямой кишки - положительный результат; в развернутой стадии (нефротической) в первом случае результаты положительные у половины больных, а во втором - еще чаще.
Наконец, при хронической почечной недостаточности данные биопсии ткани десны положительны более чем в половине наблюдений, а слизистой оболочки прямой кишки почти во всех случаях. Следовательно, биопсию слизистой оболочки десны следует рекомендовать при далеко зашедшем процессе, а прямой кишки - в любой стадии амилоидоза.
При подозрении на идиопатический амилоидоз (протекает чаще с поражением сердца, периферических нервов, реже - почек) целесообразно прежде всего проводить биопсию слизистой оболочки десны, а при вторичном (приобретенном) амилоидозе и наследственных его формах (протекают с преимущественным поражением почек) - биопсию слизистой оболочки прямой кишки.
Ряд других исследований помогает: 1) уточнить диагноз заболевания, на фоне которого развился амилоидоз; 2) оценить функциональное состояние почек (проба Реберга, Зимницкого, уровень креатинина крови).
Течение. Клиническая картина амилоидоза почек имеет особенности, отличающие его от поражения почек иного происхождения: 1) неф-ротический синдром развивается постепенно и нередко после длительной стадии протеинурии, отличается упорным течением, отеки часто резистентны к различным мочегонным средствам. При ХГН нефротический синдром возникает, как правило, уже в начале болезни и в дальнейшем часто рецидивирует; 2) артериальная гипертензия наблюдается нечасто, даже в стадии хронической почечной недостаточности; 3) при первичном амилоидозе хроническая почечная недостаточность протекает более доброкачественно в отличие от вторичного амилоидоза или ХГН (вследствие меньшей тяжести поражения клубочков по сравнению со вторичными формами амилоидоза); 4) течение вторичного амилоидоза в значительной степени зависит от основного заболевания, при частых обострениях которого возможно значительное прогрессирование амилоидоза.
Осложнения. При амилоидозе в 2 -5 % случаев развиваются:
1) тромбоз почечных вен (при вторичном амилоидозе), что проявля
ется гематурией и болями в поясничной области, нарастанием протеину
рии и падением диуреза;
2) интеркуррентная инфекция;
3) фибринозно-гнойный перитонит, появление которого сопровожда
ется резким увеличением асцита.
Диагностика. Клинические проявления амилоидоза неспецифичны. Каждый из симптомов (отеки, протеинурия, артериальная гипертензия) могут встречаться при различных заболеваниях почек. Единственным методом достоверной диагностики амилоидоза является биопсия органа (почка, печень, слизистая оболочка прямой кишки или десны), однако она не всегда выполнима. Поэтому в большинстве случаев приходится ориентироваться на клинические проявления патологического процесса.
Наличие заболевания, при котором может развиться вторичный
амилоидоз (клинические или анамнестические признаки).
Появление и прогрессирование протеинурии или возникновение
нефротического синдрома.
Заболевание, при котором может развиться амилоидоз, отсутствует,
однако имеется протеинурия или нефротический синдром.
Наличие стойкой тяжелой сердечной недостаточности, синдрома не
достаточности всасывания, полинейропатии (если при этом послед
ние три синдрома трудно объяснить другими причинами).
Можно предположить наличие амилоидоза при следующих лабораторных признаках нефротического синдрома (который, как известно, может развиваться и при других заболеваниях почек):
а) выраженная диспротеинемия + гипоальбуминемия + гипер-ссг- и ги-
пергаммаглобул инемия;
б) повышение уровня аг-гликопротеида, р-липопротеидов;
в) появление в моче а- и особенно у-гликопротеидов и а-липопро-
теидов.
Во всех случаях вероятность развития амилоидоза увеличивается при обнаружении гепато- и спленомегалии, а также изменений сердца, свойственных амилоидозу (в подобных случаях речь идет об идиопатическом генерализованном амилоидозе).
Следовательно, диагноз амилоидоза почек с достаточной уверенностью можно поставить в развернутой (нефротической) или терминальной стадии, тогда как в начальной (протеинурической) стадии это сделать много труднее. В этих случаях преходящую или постоянную протеинурию приходится дифференцировать от гломерулонефритов (острого, хронического). При этом следует учитывать:
1) более медленное прогрессирование поражения почек при амилои
дозе;
2) отсутствие при амилоидозе четкой связи с простудными заболева
ниями;
3) постоянное наличие при гломерулонефритах микрогематурии (при
амилоидозе в 20 % случаев).
Иногда правильный диагноз можно поставить лишь после периода длительного наблюдения за больным. Вопрос решается значительно быстрее, если имеется возможность произвести пункционную биопсию почки.
Формулировка развернутого клинического диагноза амилоидоза учитывает следующие компоненты: 1) форму амилоидоза; 2) стадию амилоидоза (протеинурическая, нефротическая, терминальная); 3) функциональное состояние почек (отсутствие или наличие почечной недостаточности, степень ее выраженности); 4) основное заболевание (при вторичном амилоидозе); 5) состояние других органов (сердце, печень, нервная система и пр.) при идиопатическом (первичном) амилоидозе.
Лечение. Все еще остается нерешенной проблема лечения амилоидоза, так как не выяснены причины, приводящие к усилению амилоидогене-за и недостаточности его резорбции. Тем не менее возможно проведение серии лечебных мероприятий, улучшающих состояние больного. В настоящее время лечение больного амилоидозом проводится с учетом: 1) воздействия на основное заболевание, на фоне которого развился амилоидоз
(вторичный); 2) воздействия на механизмы патогенеза; 3) воздействия на основные клинические синдромы.
Воздействие на основное заболевание, на фоне которого развивает
ся вторичный амилоидоз, необходимо ввиду того, что частые обо
стрения или высокая активность патологического процесса ведут к
прогрессированию амилоидоза.
Это воздействие заключается в следующем:
а) при хронических инфекциях (туберкулез, сифилис) необходима
длительная специфическая терапия;
б) при хронических неспецифических заболеваниях легких - ком
плексная терапия с применением антибиотиков, бронхиального дренажа, а
при необходимости и оперативное вмешательство (например, при хрони
ческом абсцессе легкого);
в) при системных заболеваниях соединительной ткани, например при
ревматоидном артрите, показана комплексная терапия, включающая на
значение базисных препаратов (D-пеницилламин, соли золота, аминохи-
нолиновые препараты).
Воздействие на механизмы патогенеза предполагает уменьшение
синтеза амилоида:
а) ежедневный прием 80-120 г сырой печени в течение 6-12 мес
приводит к снижению протеинурии, уменьшению размеров пече
ни и селезенки;
б) аминохинолиновые препараты (хингамин, или делагил, по
0,25 - 0,5 г в день в течение многих месяцев и даже лет) снижа
ют прогрессирование процесса. По-видимому, эти средства влия
ют на синтез амилоидных фибрилл. Лечение эффективно только
в ранних стадиях амилоидоза; при далеко зашедшем процессе
(развернутый нефротический синдром, почечная недостаточ
ность) назначение этих препаратов нецелесообразно;
в) при развитии амилоидоза вследствие периодической болезни ре
комендуется колхицин;
г) при первичном амилоидозе назначают также мелфалан, угнетаю
щий функцию некоторых клонов лимфоцитов, в частности син
тезирующих легкие цепи иммуноглобулинов, участвующих в
формировании амилоидной фибриллы (это имеет отношение и к
амилоидозу, развивающемуся при миеломной болезни).
Воздействие на основные клинические синдромы предусматривает
ликвидацию отеков, АГ, а также мероприятия, направленные на
борьбу с развивающейся почечной недостаточностью:
а) при развитии нефротического синдрома и выраженных отеков
необходимо достаточное содержание в пище белка, снижение по
варенной соли, а также введение цельной крови или эритроцит-
ной массы (особенно при наличии анемии), осторожное приме
нение мочегонных средств;
б) артериальная гипертензия нечасто встречается при амилоидозе,
однако, когда она достигает высоких цифр, необходимо назначе
ние гипотензивных средств различного типа;
в) при развитии почечной недостаточности лечение проводится по общепринятому плану (ограничение белка в пище, достаточное введение жидкости, коррекция минерального обмена). При почечной недостаточности, обусловленной амилоидозом, возможно применение гемодиализа и трансплантации почек.
Прогноз. Длительность протеинурического периода установить трудно, однако после его выявления обычно через 3 года развиваются отеки, на фоне которых быстро возникает ХПН. Все это делает прогноз достаточно серьезным.
Профилактика. При идиопатическом и генетическом амилоидозе меры первичной профилактики неизвестны. При вторичном амилоидозе профилактика состоит в лечении заболеваний, ведущих к развитию амилоид оза.
11160 0
Амилоидоз - это редкое заболевание, при котором в органах и тканях больного накапливается аномальный протеин (белок), называемый амилоидным белком.
В результате отложения этого белка нарушается структура и функции пораженных тканей.
Амилоидоз - это серьезная угроза для здоровья, которая может приводить к отказу жизненно важных органов и к смерти больного.
Типы амилоидоза.
Многие типы белков могут приводить к формированию амилоидных отложений, но только несколько из них связаны с тяжелым поражением органов. Тип амилоидного белка и место его накопления определяет тип амилоидоза, которым болен человек.Амилоидные отложения могут появляться в отдельных органах, либо по всему организму.
Существуют следующие типы амилоидоза:
1. Первичный (системный AL) амилоидоз. Заболевание возникает по непонятной причине, но оно часто наблюдается у больных множественной миеломой (рак крови). Это наиболее частая форма амилоидоза. Понятие «системный» означает, что амилоидоз поражает весь организм. Чаще всего поражаются почки, сердце, печень, кишечник и некоторые нервы. Форма AL вызывается т.н. «амилоидом легких цепей» (тип протеина).
2. Вторичный (системный АА) амилоидоз. Этот тип является результатом других хронических заболеваний, таких как ревматоидный артрит, волчанка, туберкулез, болезнь Крона, язвенный колит и некоторые виды рака. Он чаще всего поражает селезенку, почки, печень, надпочечники и лимфатические узлы. АА - это тип протеина, вызывающего болезнь.
3. Семейный, или наследственный ATTR-амилоидоз (AF). Эта редкая форма болезни передается по наследству. ATTR означает «амилоидный транстиретиновый белок», который и отвечает за возникновение семейного амилоидоза.
Некоторые формы амилоидных отложений, как показывают современные западные исследования, также связаны с болезнью Альцгеймера. Тем не менее, мозг довольно редко поражается амилоидозом.
Факторы риска амилоидоза.
Установлено, что мужчины болеют амилоидозом чаще женщин. Риск амилоидоза повышается о мере старения человека.Амилоидоз чаще развивается у больных с последней стадией заболеваний почек, которые находятся на диализе длительное время. Это явление вызвано накоплением бета-2-микроглобулина в крови. Диализ-индуцированный амилоидоз более характерен для взрослых больных, которые находятся на диализе более 5 лет.
Симптомы амилоидоза.
Симптомы амилоидоза часто трудно определить. Они могут очень отличаться, в зависимости от типа амилоидного белка и места его отложения в теле. Важно то, что симптомы, приведенные ниже, могут наблюдаться при многих других заболеваниях. Дифференцировать амилоидоз - нелегкая задача для квалифицированных врачей.Симптомы амилоидоза могут включать:
1. Изменения цвета кожи.
2. Глинистая окраска стула.
3. Повышенная утомляемость.
4. Общая слабость.
5. Чувство тяжести в животе.
6. Боли в суставах.
7. Анемия.
8. Одышка.
9. Отечность языка.
10. Онемение конечностей.
11. Слабое сжатие руки.
12. Потеря веса.
Сердечный амилоидоз .
Амилоидный белок откладывается в сердечной мышце. Это нарушает эластичность тканей, ослабляет сердечные сокращения и влияет на ритм сердца.
Заболевание проявляется сердечной недостаточностью (СН) - сердце не в состоянии качать объемы крови, необходимые для нормального кровоснабжения тела.
Эта форма амилоидоза проявляется такими симптомами:
1. Одышка, даже в покое.
2. Нерегулярное сердцебиение (аритмия).
3. Признаки СН - отечность, слабость, тошнота и др.
Почечный амилоидоз.
Почки должны фильтровать токсины из крови. Амилоидные отложения в почках затрудняют функционирование почек. Когда падает фильтрующая способность почек, жидкость и опасные токсины накапливаются в организме (почечная недостаточность).
При почечном амилоидозе могут быть такие симптомы:
1. Отеки, вызванные накоплением жидкости.
2. Высокий уровень белка в моче (протеинурия).
Желудочно-кишечный амилоидоз.
Амилоидный белок накапливается в желудочно-кишечном тракте, замедляя мышечные сокращения и работу кишечника. Это ухудшает пищеварение.
Если амилоид накапливается в ЖКТ, это может проявляться такими симптомами:
1. Плохой аппетит.
2. Понос (диарея).
3. Тошнота и рвота.
4. Боль в желудке.
5. Потеря веса.
Вовлечение печени может приводить к увеличению печени, накоплению жидкости в организме и аномальными изменениями в печеночных тестах.
Амилоидная нейропатия.
Амилоид может повреждать нервы, идущие от спинного и головного мозга к органам (периферические нервы). Периферические нервы несут сигнал от ЦНС к различным участкам тела. К примеру, периферические нервы дают возможность чувствовать зуд в кончике пальца, или ожог ладони.
Если амилоидоз поражает периферические нервы, то это проявляется такими симптомами:
1. Проблемы с равновесием.
2. Нарушение контроля над мочеиспусканием и дефекацией.
3. Нарушение выделения пота.
4. Покалывание и слабость в мышцах.
5. Головокружение в положении стоя, вызванное проблемами с регуляцией артериального давления.
Кроме всего, сказанного выше, амилоидоз может также поражать легкие, кожу, селезенку и другие органы, вызывая соответствующие симптомы.
Диагностика амилоидоза.
Врач должен провести внимательный физический осмотр и изучить историю болезни, чтобы заметить подозрительные признаки, которые могут указывать на амилоидоз. Как уже говорилось, эти признаки не специфичные, и могут говорить о многих других заболеваниях.Не существует определенного анализа крови, который позволяет выявить амилоидоз. Тонкая лабораторная методика, называемая электрофорезом свободных легких цепей, позволяет выявить ранние признаки наличия некоторых амилоидных протеинов.
Стоит ли говорить, что далеко не все медицинские учреждения способны провести такие анализы?
Биопсия необходима для того, чтобы подтвердить диагноз и определить специфический тип амилоидного белка, который вызвал болезнь. Образец ткани можно взять изо рта, прямой кишки, внутренних органов.
При подозрении на наличие семейного (наследственного) амилоидоза, врач может назначить генетический анализ. Лечение наследственного амилоидоза будет зависеть от особенностей болезни.
Также врач может назначить множество анализов крови, мочи, сканирование тела с целью определить повреждения тех или иных органов и тканей в результате болезни.
Лечение амилоидоза.
Не существует радикального лечения амилоидоза.Врач может назначить лечение для подавления выработки амилоидного белка, а также поддерживающую терапию для нормализации функций поврежденных органов. Если амилоидоз связан с другим заболеванием, то лечение должно быть нацелено и на это заболевание.
Конкретная схема лечения будет зависеть от типа амилоидоза и органов, которые вовлечены в патологический процесс.
Возможные варианты лечения:
1. Лечение стволовыми клетками помогает удалить вещества, которые приводят к накоплению амилоида у больных первичным AL-амилоидозом, у которых повреждено не более двух важных органов.
2. Химиотерапевтические средства используются для лечения остальных пациентов с первичным AL-амилоидозом.
3. Мощные противовоспалительные средства (кортикостероиды) используются для лечения вторичного AA-амилоидоза.
4. Пересадка печени может остановить болезнь у пациентов с наследственной формой амилоидоза.
5. Пересадка почек и сердца может быть рекомендована при серьезном повреждении этих жизненно важных органов.
Другие методы борьбы с симптомами амилоидоза могут включать:
1. Мочегонные средства для удаления избытка жидкости из организма.
2. Компрессионные чулки для облегчения отека нижних конечностей.
3. Специальная диета, особенно при желудочно-кишечном амилоидозе.
Прогноз амилоидоза.
Амилоидоз может быть смертелен, особенно в случае поражения почек или сердца. Ранняя диагностика и правильное лечение очень важны для повышения выживаемости. При отсутствии лечения многие пациенты умирают в течение двух лет после постановки диагноза.Исследователи продолжают изучать, почему некоторые типы вызывают болезнь, и как можно остановить формирование амилоидных белков. Ведутся масштабные научные работы по созданию новых лекарств от амилоидоза.
На Западе многие, казалось бы, безнадежные больные могут получить возможность участвовать в клинических испытаниях новейших препаратов, причем многие из них помогают продлить жизнь таким больным.
Константин Моканов
Кандидат медицинских наук В.Н. Кочегуров
АМИЛОИДОЗ ВНУТРЕННИХ ОРГАНОВ
В последние годы изменились многие представления об амилоидозе и методах его лечения. Под названием «амилоидоз» объединяется группа заболеваний, отличительным признаком которых является отложение в тканях особого гликопротеида, состоящего из фибриллярных или глобулярных белков, тесно связанных с полисахаридами, с нарушением структуры и функции пораженных органов.
Термин «амилоид» ввел в 1854 г. Р. Вирхов, который подробно изучил вещество, откладывающееся в тканях при так называемой сальной болезни у лиц, страдающих туберкулезом, сифилисом, актиномикозом, и счел его похожим на крахмал из-за характерной реакции с йодом. И только через 100 лет Кохеном с помощью электронной микроскопии была установлена его белковая природа.
Амилоидоз - достаточно распространенная патология, особенно если учесть существование его локальных форм, частота которых значительно увеличивается с возрастом.
Многообразие форм и вариантов амилоидоза не дает возможности систематизировать сведения об этиологии и патогенезе.
Современная классификация амилоидоза построена по принципу специфичности основного белка, образующего амилоид. По классификации ВОЗ (1993), вначале приводится тип амилоида, затем указывается белок-предшественник и уже потом - клинические формы заболевания с перечислением преимущественных органов-мишеней. Во всех названиях типов амилоида первой буквой является «А», означающая «амилоид», далее за ней следует аббревиатура конкретного фибриллярного белка, из которого он образовался:
АА-амилоидоз . Второе «А» - это обозначение острофазового белка (SSA--глобулин), продуцируемого в ответ на воспаление или наличие опухоли (anacute-phaseprotein);
AL -амилоидоз. «L» - это легкие цепи иммуноглобулинов (lightchains);
ATTR -амилоидоз. «TTR» - это транстиретин, транспортный белок для ретинола и тироксина;
A 2 М-амилоидоз. « 2 М» - это 2 -микроглобулин (диализный амилоидоз).
АА-амилоидоз. АА-амилоид образуется из сывороточного острофазового белка, представляющего собой-глобулин, который синтезируется гепатоцитами, нейтрофилами и фибробластами. Его количество повышается многократно при воспалении или наличии опухолей. Однако участвуют в образовании амилоида лишь отдельные его фракции, поэтому амилоидоз развивается лишь у части пациентов с воспалительными или опухолевыми заболеваниями. Конечный этап амилоидогенеза – полимеризация растворимого предшественника в фибриллы – до конца не выяснен. Считается, что этот процесс происходит на поверхности макрофагов при участии мембранных ферментов и тканевых факторов, что и определяет органное поражение.
АА-амилоидоз объединяет 3 формы:
Вторичный реактивный амилоидоз при воспалительных и опухолевых заболеваниях. Это самая распространенная форма. В последние годы среди причин вторичного амилоидоза на первый план вышли ревматоидный артрит, болезнь Бехтерева, псориатический артрит и опухоли, в т.ч. системы крови (лимфома, лимфогранулематоз), а также неспецифический язвенный колит и болезнь Крона. В то же время хронические гнойно-обструктивные заболевания легких отступают на второй план, так же как туберкулез и остеомиелит.
Периодическая болезнь (семейная средиземноморская лихорадка) с аутосомно-рецессивной формой наследования. Отмечается этническая предрасположенность к ней у арабов, армян, евреев и цыган. Выделяют 4 формы этого заболевания: лихорадочную, суставную, торакальную и абдоминальную. На первом-втором десятилетиях жизни у больных возникают немотивированная лихорадка или проявления артрита. Дебют болезни возможен с развития клиники сухого плеврита или картины «острого» живота. Причем эти эпизоды обычно кратковременные, длительностью 7-10 дней, стереотипны по своим проявлениям и длительное время не вызывают осложнений (деформаций и дефигураций суставов, спаек или шварт плевральных листков, спаечной болезни брюшной полости). Однако у 40% больных на втором-третьем десятилетиях жизни развивается прогрессирующий амилоидоз почек.
Синдром Макла-Уэльса или семейная нефропатия с крапивницей и глухотой, наследуемый по аутосомно-доминантному типу. В первые годы жизни у больных периодически возникают аллергические сыпи, чаще в виде крапивницы или отека Квинке, сопровождающиеся лихорадкой, лимфаденопатией, артро- и миалгиями, болями в животе, эозинофильными инфильтратами в легких. Эти симптомы спонтанно исчезают через 2-7 суток с последующей ремиссией. Параллельно возникает и прогрессирует снижение слуха, а на втором-третьем десятилетиях жизни присоединяется амилоидоз почек. Это наиболее распространенный вариант наследственного амилоидоза.
Органами-мишенями при АА-амилоидозе чаще всего бывают почки, а также печень, селезенка, кишечник и надпочечники.
А L -амилоидоз . АL-амилоид образуется из легких цепей иммуноглобулинов, в которых изменена последовательность аминокислот, вызывающая дестабилизацию этих молекул и способствующая образованию фибрилл амилоида. В этом процессе участвуют местные факторы, особенности которых определяют поражение тех или иных органов. Иммуноглобулины синтезируются аномальным клоном плазматических или В-клеток в костном мозге, появившихся, по-видимому, в результатемутации или Т-иммунодефицита и снижения контролирующей функции последних.
АL-амилоидоз включает в себя 2 формы:
1) Первичный идиопатический амилоидоз , при котором нет предшествующего заболевания;
2) Амилоидоз при миеломной болезни и В-клеточных опухолях (болезнь Вальденстрема, Франклина и др.). АL-амилоидоз сейчас рассматривают в рамках единой В-лимфоцитарной дискразии.
К основным органам-мишеням при АL-амилоидозе относятся сердце, желудочно-кишечный тракт, а также почки, нервная система, кожа. Дефицит Х фактора свертывания крови при АL-амилоидозе считают причиной развития геморрагического синдрома с характерными кровоизлияниями вокруг глаз («глаза енота»).
В дифференциальной диагностике системного амилоидоза следует принимать во внимание то, что АА-тип более «молодой», средний возраст заболевших составляет менее 40 лет, а при АL-амилоидозе - 65 лет, причем при обоих типах отмечается преобладание мужчин (1,8-1).
ATTR -амилоидоз включает в себя 2 варианта:
Семейная нейропатия (реже кардио- и нефропатия) с аутосомно-доминантным типом наследования. При этомATTR-амилоид образуется измутантного транстиретина, синтезируемого гепатоцитами. Мутантные белки нестабильны и при определенных условиях преципитируют в фибриллярные структуры, образуя амилоид.
Системный старческий амилоидоз , развивающийся исключительно у пожилых людей (старше 70 лет). В его основе лежит нормальный по аминокислотному составу транстиретин (т.е. не мутантный), но с измененными физико-химическими свойствами. Они связаны с возрастными метаболическими сдвигами в организме и обусловливают образование фибриллярных структур.
Для этого варианта характерно поражение нервной системы, реже почек и сердца.
A 2 М-амилоидоз – это относительно новая форма системного амилоидоза, появившаяся в связи с внедрением в практику хронического гемодиализа. Белком-предшественником является 2 -микроглобулин, который не фильтруется при гемодиализе через большинство мембран и задерживается в организме. Его уровень повышается в 20-70 раз, что служит основой для развития амилоидоза в среднем через 7 лет от начала гемодиализа.
Основными органами-мишенями являются кости и периартикулярные ткани. Могут возникнуть патологические переломы костей. В 20% случаев наблюдается синдром карпального канала (онемение и боль в первых трех пальцах кисти, распространяющиеся на предплечье с последующим развитием атрофии мышц тенара из-за сдавления срединного нерва отложениями амилоидных масс в области карпальной связки).
Кроме системных форм выделяют локальный амилоидоз , который возникает в любом возрасте, но чаще в пожилом, и поражает любую ткань или орган. Практическое значение имеет амилоидоз островков поджелудочной железы у стариков (ААIАРР-амилоид). Сейчас накоплено достаточно фактов, свидетельствующих о том, что почти все случаи диабета 2 типа у людей пожилого возраста патогенетически связаны с амилоидозом островкового аппарата поджелудочной железы, который образуется из полипептида-клеток.
Церебральный амилоидоз (АВ-амилоид) рассматривают как основу церебральной деменции Альцгеймера. При этом сывороточный-протеин откладывается в старческих бляшках, мозговых нейрофибриллах, сосудах и оболочках.
Среди всех типов амилоидоза наибольшее значение имеют АА- и АL-формы системного амилоидоза.
Амилоидоз почек. Наиболее часто поражаемым органом при системном амилоидозе являются почки. Сначала амилоид откладывается в мезангиуме, потом вдоль базальной мембраны клубочков, пенетрируя в нее и вскрывая подэпителиальное пространство и камеру Шумлянского-Боумена. Затем амилоид откладывается в стенках сосудов, строме пирамид, капсуле почек.
Первым клиническим проявлением амилоидоза почек является протеинурия , которая зависит не столько от величины отложений амилоида, сколько от деструкции клеток-подоцитов и их ножек. Сначала она носит транзиторный характер, иногда сочетается с гематурией или/и лейкоцитурией. Этолатентная стадия нефропатического варианта амилоидоза. С момента стабилизации протеинурии наступает вторая –протеинурическая стадия. По мере увеличения протеинурии и формирования гипопротеинемии с развитием вторичного альдостеронизма и возникновением нефротических отеков наступает третья –нефротическая стадия . При снижении функций почек и появлении азотемии наступает четвертая –азотемическая стадия поражения почек.
В «классических» случаях у больных амилоидозом почек формируется нефротический синдром (НС) с его отечным периодом, причем время развития НС индивидуально. Важно отметить, чтоартериальная гипертензия не является характерным признаком , так как происходит поражение ЮГА со снижением продукции ренина, и она может возникнуть лишь у 10-20% больных при далеко зашедшей ХПН.
Примечательно, что при амилоидозе размеры почек сохраняются неизмененными или даже увеличиваются («большие сальные почки» ), несмотря на нарастание их функциональной неполноценности. Выявление этого симптома с помощью УЗ–сканирования и рентгенологического метода является важным диагностическим критерием амилоидного поражения почек.
Сердце при амилоидозепоражается часто, особенно при АL-варианте. В результате отложения амилоида в миокарде нарастает ригидность сердечной стенки, страдает функция диастолического расслабления.
Клинически это проявляется кардиомегалией (вплоть до развития «бычьего сердца»),глухостью тонов, прогрессирующей рефрактерной к лечению сердечной недостаточностью , которая является причиной смерти у 40% больных. У части больных развивается инфаркт миокарда вследствие отложений амилоида в коронарных сосудах, стенозирующих их просвет. Возможно вовлечение клапанов сердца с развитием того или иного порока сердца и поражения перикарда, напоминающего констриктивный перикардит.
На ЭКГ регистрируется снижение вольтажа зубцов, при эхокардиографии отмечается симметричное утолщение стенок желудочков с признаками диастолической дисфункции. В зависимости от локализации депозитов амилоида в миокарде может наблюдаться синдром слабости синусового узла, АВ-блокада, разнообразные аритмии, иногда очаговые поражения с инфарктоподобной картиной на ЭКГ.
Желудочно-кишечный тракт при амилоидозепоражается на всем протяжении.Макроглоссия, встречающаяся у 22% больных с амилоидозом, являетсяпатогномоничным симптомом . При этом развиваетсядисфагия, дизартрия, глоссит, стоматит , а по ночам не исключена асфиксия вследствие западения языка и перекрытия им дыхательных путей.
Отложение амилоида в пищеводе сопровождается нарушениями его функций, иногда находятопухолевидные образования в желудке и кишечнике . Нередко поражается мышечный слой кишечника и нервные сплетения, что приводит к нарушению моторики ЖКТ, вплоть до возникновенияилеуса . Отложение амилоида в тонком кишечнике приводит к появлениюсиндромов мальабсорбции и мальдигестии . Вследствие поражения сосудов образуютсяязвы кишечника с развитием кровотечений, что симулирует картину опухолей или неспецифического язвенного колита.
Отложение амилоида в поджелудочной железе приводит к ее внешне- и внутрисекреторной недостаточности.
С большой частотой вовлекается в процесс печень (у 50% больных АА-амилоидозом и у 80% - АL-амилоидозом). Характерно длительное сохранение функций печени сотсутствием синдромов цитолиза и холестаза . В развернутой стадии появляютсяпризнаки портальной гипертензии с кровотечениями из варикозно расширенных вен. Типичнажелтуха за счет сдавления желчных капилляров. Часто определяетсяспленомегалия с явлениями гиперспленизма , а такжеувеличение периферических лимфоузлов .
Дыхательная система чаще всего вовлекается в процесс при АL-амилоидозе (у 50% больных), реже - при АА-амилоидозе (10-14%).
К ранним признакам относится осиплость голоса , связанная с отложением амилоида в голосовых связках. Затем присоединяется поражение бронхов, альвеолярных перегородок, сосудов. Возникаютателектазы и инфильтраты легких, диффузные изменения по типу фиброзирующего альвеолита с дыхательной недостаточностью и легочной гипертензией, способствующие формированиюхронического легочного сердца . Возможны легочные кровотечения или развитие локального легочного амилоидоза, имитирующего картину рака легкого.
Вовлечение периферической и вегетативной нервной системы отмечается при системном амилоидозе разных типов, но в большей степени при АL- и АТТR-типах. Периферическая сенсорная, иногда моторная нейропатия (как правило, симметричная, начинающаяся с дистальных отделов конечностей и распространяющаяся на проксимальные) может преобладать в клинической картине, создавая диагностические трудности. Нарушения вегетативной нервной системы могут быть значительно выражены и проявляются симптомами ортостатической гипотонии, импотенцией, сфинктерными расстройствами.
Центральная нервная система при амилоидозе поражается редко.
Среди поражений других органов следует отметить возможность поражения надпочечников и щитовидной железы с развитием симптомов их недостаточности.
Амилоидные отложения в коже могут иметь вид папул, узлов, бляшек, диффузной ее инфильтрации с трофическими изменениями, приобретенного тотального альбинизма.
Вовлечение в процесс суставов и периартикулярных тканей , как уже упоминалось, связано с диализным амилоидозом.
Поражение скелетных мышц обычно резко снижает качество жизни больных. Сначала отмечается псевдогипертрофия мышц с последующей их атрофией, приводящей к обездвиживанию больного.
Изменение лабораторных показателей при амилоидозе неспецифично: увеличение СОЭ, гиперглобулинемия, тромбоцитоз, который наряду с малыми размерами тромбоцитов и появлением эритроцитов с тельцами Жолли рассматривают как свидетельствогиперспленизма .
Диагностика амилоидоза, предполагаемого по клиническим признакам, должна быть подтверждена обнаружением субстрата патологии, а именно, амилоида.
С этой целью можно использовать красочные пробы . В одной из модификаций больному внутривенно вводят краситель (синька Эванса, конго-красный ), который может захватываться амилоидными массами, что приводит к снижению его концентрации в крови.
В другом варианте исследования больному подкожно в подлопаточную область вводят 1 см 3 1% свежеприготовленного раствораметиленового синего и затем следят за изменением цвета мочи. Если амилоидные массы захватили краситель, цвет мочи не изменяется, и проба считается положительной, что подтверждает диагноз амилоидоза. Если проба отрицательная (цвет мочи изменен), то это не исключает наличие амилоидоза.
Другим методом диагностики является биопсия. Если производится биопсия пораженного органа (почки, печени и др.), то частота положительных результатов достигает 90-100%. Чем выше степень инфильтрации органов-мишеней амилоидом, тем больше возможность его обнаружения. Обычно диагностику амилоида начинают с биоптатов слизистой оболочки полости рта с подслизистым слоем в области десны около 3-4 моляра или в прямой кишке. При АL-амилоидозе рекомендуют в первую очередь проводить биопсию костного мозга или аспирационную биопсию подкожно-жировой клетчатки передней брюшной стенки (чувствительность около 50%). При диализном амилоидозе целесообразна биопсия периартикулярных тканей.
В последние годы все шире применяется сцинтиграфия с меченымI 123 сывороточным Р-компонентом для оценкеinvivoраспределения амилоида в организме. Метод особенно полезен для контроля динамики его тканевых отложений в процессе лечения. Важно не только обнаружить амилоид в тканях, но и провести его типирование с помощью красочных методов или, что более точно, с помощью антисывороток (поли- и моноклональные антитела) к основным белкам амилоидных фибрилл.
Лечение амилоидоза должно быть направлено на уменьшение синтеза и доставки белков-предшественников, из которых строится амилоид.
При лечении АА-амилоидоза , его вторичного варианта, необходимым условием является терапия заболевания, приведшего к развитию амилоидоза всеми доступными методами (антибиотики, химиопрепараты, хирургическое вмешательство).
Препаратами выбора являются 4-аминохинолиновые производные (делагил, плаквенил, резохин, хингамин и др.). Они тормозят синтез амилоидных фибрилл на ранних стадиях амилоидогенеза, ингибируя ряд ферментов. Делагил назначают по 0,25 г длительно (годами).
Белковые фибриллы, образующие амилоид, содержат большое количество свободных сульфгидрильных групп (SH), которые активно участвуют в агрегации белков в стабильные структуры. С целью их блокады применяютунитиол по 3-5 мл 5% раствора внутримышечно ежедневно с постепенным увеличением дозы до 10 мл в сутки в течение 30-40 дней и повторными курсами 2-3 раза в год.
По-прежнему рекомендуется прием сырой или кулинарно обработанной печени по 100-150 г в сутки в течение 6-12 месяцев. Белки и антиоксиданты печени тормозят развитие амилоидоза. Можно использовать такжепеченочные гидролизаты , в частностисирепар (2 мл сирепара соответствуют 40 г печени), и проводить лечение, чередуя прием сырой печени в течение 1-2 месяцев с 2-3 месячным применением сирепара (внутримышечно по 5 мл 2 раза в неделю).
Применяют иммуномодуляторы: левамизол (декарис) по 150 мг 1 раз в 3 дня (2-3 недели), тималин по 10-20 мг внутримышечно 1 раз в день (5 дней), Т-активин по 100 мкг внутримышечно 1 раз в день (5 дней).
Признается положительный эффект димексида , оказывающего прямое рассасывающее действие. Его применяют перорально в виде 10-20% раствора в суточной дозе не менее 10 г в течение 6 месяцев.
При периодической болезни показанколхицин , обладающий антимитотическим действием. Препарат замедляет амилоидогенез. Его раннее назначение может предупредить возникновение амилоидоза почек, что является наиболее опасным при данной патологии. Назначают длительно (пожизненно) в дозе 1,8-2 мг в сутки (таб. 2 мг).
Лечение А L -амилоидоза . Поскольку этот тип амилоидоза рассматривают в рамках моноклональной пролиферации плазматических или В-клеток, то в лечении применяют различные схемыполихимиотерапии с целью уменьшить продукцию предшественников – легких цепей иммуноглобулинов. Чаще всего используют схему цитостатикмелфолан+преднизолон (мелфолан в дозе 0,15 мг/кг, преднизолон по 0,8 мг/кг по 7 дней каждые 4-6 недель в течение 2-3 лет). Сейчас применяют и более агрессивные схемы с включением винкристина, доксорубицина, циклофосфана.
Существует мнение о целесообразности применения левамизола или других иммуномодуляторов для повышения функции Т-супрессоров.
В лечении АТТ R -амилоидоза наиболее эффективнатрансплантация печени .
Для лечения A 2 М- илидиализного амилоидоза применяют высокопоточный гемодиализ с гемофильтрацией и иммуносорбцией. Благодаря этому снижается уровень 2 -микроглобулина. При необходимости производяттрансплантацию почек .
Следует отметить, что проведение адекватного лечения часто оказывается невозможным из-за позднего распознавания болезни с вовлечением в патологический процесс многих органов. Поэтому решающее значение имеет ранняя диагностика, основанная на знании разнообразных проявлений амилоидоза.
Профилактика. Основной профилактики вторичного амилоидоза является успешное лечение гнойно-воспалительных, системных и опухолевых заболеваний. В случаях идиопатического амилоидоза проблема профилактики должна решаться путем тщательного сбора анамнеза семейно-наследственных заболеваний и медико-генетического консультирования.
 Амилоидоз (амилоидная дистрофия, лат. amyloidosis, греч. amylon крахмал + eidos вид + ōsis) – группа заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным (во внеклеточном матриксе) отложением (системным или локальным) нерастворимых патологических фибриллярных белков (белково-полисахаридного комплекса – амилоида) в органах и тканях, которые образуются в результате сложных обменных изменений (белковых дистрофий). Основными органами-мишенями являются сердце, почки, нервная система [центральная и периферическая], печень, однако при системных формах могут поражаться практически все ткани (к редким локализациям относят амилоидоз надпочечников). Амилоидами их назвали потому что, в реакции с йодом они напоминали крахмал. Амилоид долгое время персистирует в организме и даже после смерти в течение долгого времени не подвергается гниению (И.В. Давыдовский, 1967). Амилоидоз может возникнуть самостоятельно или «вторично» в результате другого заболевания.
Амилоидоз (амилоидная дистрофия, лат. amyloidosis, греч. amylon крахмал + eidos вид + ōsis) – группа заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным (во внеклеточном матриксе) отложением (системным или локальным) нерастворимых патологических фибриллярных белков (белково-полисахаридного комплекса – амилоида) в органах и тканях, которые образуются в результате сложных обменных изменений (белковых дистрофий). Основными органами-мишенями являются сердце, почки, нервная система [центральная и периферическая], печень, однако при системных формах могут поражаться практически все ткани (к редким локализациям относят амилоидоз надпочечников). Амилоидами их назвали потому что, в реакции с йодом они напоминали крахмал. Амилоид долгое время персистирует в организме и даже после смерти в течение долгого времени не подвергается гниению (И.В. Давыдовский, 1967). Амилоидоз может возникнуть самостоятельно или «вторично» в результате другого заболевания.
В настоящее время амилоидоз рассматривают как группу заболеваний, которые характеризуются отложением в тканях и органах фибриллярного белка амилоида (ФБА) - особой белковой структуры диаметром 5 - 10 нм и длиной до 800 нм, состоящей из 2 и более параллельных разнонаправленных (антипараллельных) филаментов, образующих кросс-β-складчатую конформацию (см. рис. слева). Именно она определяет специфическое оптическое свойство амилоида – способность к двойному лучепреломлению (выявляемого при окраске конго красным [= метод определения амилоида в тканях]). По современным данным распространённость амилоидоза в популяции колеблется от 0,1 до 6,6%.

Название белка «амилоид» было предложено Рудольфом Вирховым, который позаимствовал его из ботаники, где это слово означало целлюлозу или крахмал. По своей структуре амилоид является сложным гликопротеидом, в котором в структуре с полисахаридами (галактозой, глюкозой, глюкозамином, галактозаминами, маннозой и фруктозой) находятся фибриллярные и глобулярные белки. В амилоиде содержатся белки, близкие по своим особенностям к α1-, β- и γ-глобулинам, альбумину, фибриногену, в нём содержится нейраминовая кислота. Связи белков и полисахаридов очень прочные, что сохраняет его стабильность. В структуре амилоида также находится Р-компонент, составляющий до 15% всего амилоида и идентичный сывороточному белку SAP (сывороточный амилоид P). SAP является белком, продуцируемым клетками печени, относящимся к категории острофазовых (SAP - постоянная составная часть амилоидных депозитов при всех формах амилоидоза).
Амилоидоз полиэтиологичен. Основное значение придают амилоидогенности основного белка-предшественника амилоида (БПА), специфичного для каждой формы амилоидоза. Амилоидогенность определяется изменениями в первичной структуре БПА, закрепленными в генетическом коде или приобретенными в течение жизни вследствие мутаций. Для реализации амилоидогенного потенциала БПА необходимо воздействие ряда факторов, таких как воспаление, возраст, физико-химические условия in situ.
ТАБЛИЦА : Классификация амилоидоза (во всех названиях типов амилоидоза первой буквой является прописная буква «А», означающая слово «амилоид», за ней следует обозначение конкретного БПА - А [амилоидный А-протеин; образуется из сывороточного белка-предшественника SAA - острофазового белка, в норме синтезируемого гепатоцитами, нейтрофилами и фибробластами в следовых количествах], L [легкие цепи иммуноглобулинов], TTR [транстиретин], 2М [β2-микро-глобулин], В [В-протеин], IAPP [островковый амилоидный полипептид] и т.д.).

Обратите внимание ! Структурные и химико-физические особенности амилоида определяются основным БПА, содержание которого в фибрилле достигает 80% и является специфичным признаком для каждого типа амилоидоза. У каждого белка (БПА) существенно отличаются механизмы синтеза, утилизации, биологические функции, что определяет различия в клинических проявлениях и подходах к лечению амилоидоза. По этой причине разные формы амилоидоза рассматривают как разные заболевания (см. таблицу ).
Несмотря на достигнутый прогресс в изучении амилоида разных типов, конечный этап амилоидогенеза - образование фибрилл амилоида в межклеточном матриксе из БПА - остается во многом невыясненным. По-видимому, это многофакторный процесс, имеющий свои особые черты при разных формах амилоидоза. Рассмотрим процесс амилоидогенеза на примере АА-амилоидоза. Считают, что при образовании АА из SAA имеют значение процесс неполного расщепления SAA протеазами, связанными с поверхностной мембраной моноцитов-макрофагов, и полимеризация растворимого АА-белка в фибриллы, происходящая, как предполагают, также при участии мембранных ферментов. Интенсивность образования АА-амилоида в тканях зависит от концентрации SAA в крови. Количество SAA, синтезируемого клетками разных типов (гепатоцитами, нейтрофилами, фибробластами), повышается во много раз при воспалительных процессах, опухолях (повышение содержания SAA в крови играет основную роль в патогенезе АА-амилоидоза). Однако для развития амилоидоза недостаточно только высокой концентрации SAA, необходимо также наличие у БПА (т.е. у SAA) амилоидогенности. Развитие амилоидоза у человека связывают с депозицией SAA1. В настоящее время известно 5 изотипов SAA1, из которых наибольшую амилоидогенность приписывают изотипам 1.1 и 1.5. Конечный этап амилоидогенеза - образование фибрилл амилоида из БПА - осуществляется при неполном расщеплении протеазами моноцитов-макрофагов. Стабилизация амилоидной фибриллы и резкое снижение растворимости этого макромолекулярного комплекса во многом обусловлены взаимодействием с полисахаридами интерстиция.
Несмотря на различие в типах амилоидного белка, существует общность патогенеза различных клинических форм амилоидоза. Основной причиной развития болезни служит наличие определенного, нередко повышенного количества амилоидогенного БПА. Появление или усиление амилоидогенности может быть обусловлено циркуляцией вариантов белков с повышенной общей гидрофобностью молекулы, нарушенным соотношением поверхностных молекулярных зарядов, что приводит к нестабильности белковой молекулы и способствует ее агрегации в амилоидную фибриллу. На последнем этапе амилоидогенеза происходит взаимодействие амилоидного белка с белками плазмы крови и гликозоаминогликанами тканей. Кроме структурных особенностей, имеют значение также физико-химические свойства межклеточного матрикса, где происходит сборка амилоидной фибриллы. Многие формы амилоидоза можно объединить также по признаку возникновения в пожилом и старческом возрасте (AL, ATTR, AIAPP, AApoA1, AFib, ALys, AANF, A-бета), что указывает на наличие механизмов возрастной эволюции структуры определенных белков в сторону повышения амилоидогенности и позволяет рассматривать амилоидоз как одну из моделей старения организма.
Неврологические аспекты амилоидоза :
ATTR-амилоидоз . К ATTR-амилоидозу относят семейную амилоидную полиневропатию, которая наследуется по аутосомно-доминантному типу, и системный старческий амилоидоз. Белком-предшественником при этой форме амилоидоза является транстиретин - компонент молекулы преальбумина, синтезируемый печенью и выполняющий функции транспортного белка тироксина. Установлено, что наследственный ATTR-амилоидоз бывает результатом мутации в гене, кодирующем транстиретин, что приводит к замене аминокислот в молекуле TTR. Существует несколько типов наследственной амилоидной нейропатии: португальский, шведский, японский и ряд других. При наиболее частом семейном варианте (португальском) в 30-й позиции от N-конца молекулы транстиретина метионин заменён на валин, что повышает амилоидогенность белка-предшественника и облегчает его полимеризацию в амилоидные фибриллы. Известно несколько вариантных транстиретинов, чем и обусловлено разнообразие клинических форм наследственной невропатии. Клинически это заболевание характеризуется прогрессирующей периферической и вегетативной невропатией, которая сочетается с поражением сердца, почек и других органов различной степени. Системный старческий амилоидоз развивается после 70 лет в результате возрастных конформационных изменений нормального транстиретина, по-видимому, усиливающих его амилоидогенность. Органы-мишени старческого амилоидоза - сердце, сосуды головного мозга и аорта.
читайте также пост: Транстиретиновая амилоидная полинейропатия (на сайт)
читайте также статью «Поражение периферической нервной системы при системном амилоидозе» Сафиулина Э.И., Зиновьева О.Е., Рамеев В.В., Козловская-Лысенко Л.В.; ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» МЗ РФ, Москва (журнал «Неврология, нейропсихиатрия, психосоматика» №3, 2018) [читать ]
Болезнь Альцгеймера (БА) - это генетически детерминированное прогрессирующее нейродегенеративное заболевание, в основе которого лежит гибель нейронов больших полушарий головного мозга; клиническими проявлениями заболевания являются снижение памяти и других когнитивных функций (интеллект, праксис, гнозис, речь). На данный момент выявлено 4 основных гена, отвечающих за развитие данного заболевания: ген, кодирующий предшественник амилоидного белка (amyloid precursor protein, APP, 21-я хромосома), гены, кодирующие ферменты [альфа-, бета-, гамма-секретазы], метаболизирующие АРР: пресенилин-1 (14-я хромосома), пресенилин-2 (1-я хромосома). Особая роль отводится гетеро- или гомозиготному носительству четвертой изоформы аполипопротеина Е (АПОЕ 4).
В норме предшественник амилоидного белка (АРР) расщепляется альфа-секретазой на растворимые (одинаковые по величине) полипептиды, которые не являются патогенными, и (АРР) выводится из организма; при патологии генов, отвечающих за метаболизм АРР, последний расщепляется бета- и гамма-секретазами на различные по длине фрагменты. При этом происходит образование нерастворимых длинных фрагментов амилоидного белка (альфа-бета-42), которые в последующем откладываются в веществе (паренхиме) головного мозга и стенках церебральных сосудов (стадия диффузного церебрального амилоидоза), что приводит к гибели нервных клеток. Далее в паренхиме головного мозга происходит агрегация нерастворимых фрагментов в патологический белок – бета-амилоид («гнездные» отложения данного белка в паренхиме головного мозга называют сенильными бляшками). Отложение амилоидного белка в церебральных сосудах приводит к развитию церебральной амилоидной ангиопатии, которая является одной из причин хронической ишемии головного мозга.

читать статью: Церебральная амилоидная ангиопатия
(на сайт)
Бета-амилоид и нерастворимые фракции диффузного амилоидного белка обладают нейротоксическими свойствами. В эксперименте показано, что на фоне церебрального амилоидоза активируются тканевые медиаторы воспаления, усиливается выброс возбуждающих медиаторов (глутамат, аспартат и др.), повышается образование свободных радикалов. Результатом всего этого сложного каскада событий является повреждение нейрональных мембран, индикатором которого является образование внутри клеток нейрофибриллярных сплетений (НФС). НФС представляют собой фрагменты биохимически измененной внутренней мембраны нейрона и содержат гиперфосфорилированный тау-протеин. В норме тау-протеин является одним из основных белков внутренней мембраны нейронов. Наличие внутриклеточных НФС свидетельствует о необратимом повреждении клетки и ее скорой гибели, после которой НФС выходят в межклеточное пространство («НФС-призраки»). В первую очередь и в наибольшей степени страдают нейроны, окружающие сенильные бляшки.

От начала отложения амилоидного белка в головном мозге до развития первых симптомов болезни - легкой забывчивости - проходит 10 - 15 лет. В значительной степени скорость прогрессии БА определяется выраженностью сопутствующей соматической патологии, сосудистых факторов риска, а также интеллектуальным развитием пациента. У пациентов с высоким уровнем образования и достаточной интеллектуальной нагрузкой заболевание течет медленнее, чем у пациентов со средним или начальным образованием и недостаточной интеллектуальной активностью. В этой связи была разработана теория когнитивного резерва, согласно которой при интеллектуальной деятельности мозг человека образовывает новые межнейрональные синапсы и в когнитивный процесс вовлекаются все большие популяции нейронов. Это облегчает компенсацию когнитивного дефекта даже при прогрессирующей нейродегенерации.
Диагностика амилоидоза . Предполагаемый на основании клинических и лабораторных данных амилоидоз необходимо подтвердить морфологически обнаружением амилоида в биоптатах тканей. При подозрении на AL-тип амилоидоза рекомендуют производить пункцию костного мозга. Наиболее часто для диагностики разных типов амилоидоза проводят биопсию слизистой оболочки прямой кишки, почки, печени. Биопсия слизистого и подслизистого слоев прямой кишки позволяет выявить амилоид у 70% больных, а биопсия почки - практически в 100% случаев. У пациентов с синдромом запястного канала исследованию на амилоид необходимо подвергать ткань, удаленную при операции декомпрессии запястного канала. Биопсийный материал для выявления амилоида необходимо окрашивать конго красным с последующей микроскопией в поляризованном свете для выявления способности к двойному лучепреломлению.
Современная морфологическая диагностика амилоидоза включает не только обнаружение, но и типирование амилоида, поскольку тип амилоида определяет терапевтическую тактику. Для типирования часто применяют пробу с перманганатом калия. При обработке окрашенных конго красным препаратов 5%-ным раствором перманганата калия АА-тип амилоида теряет окраску и утрачивает свойство двойного лучепреломления, тогда как AL-тип амилоида сохраняет их. Использование щелочного гуанидина позволяет более точно дифференцировать АА- и AL-амилоидоз. Наиболее эффективным методом типирования амилоида служит иммуногистохимическое исследование с применением антисывороток к основным типам амилоидного белка (специфические антитела против АА-белка, легких цепей иммуноглобулинов, транстиретина и бета-2-мик- роглобулина).
Обратите внимание ! Амилоидоз - полисистемное заболевание, поражение только одного органа наблюдается редко. Если в анамнезе упоминается о сочетании таких симптомов, как общая слабость, исхудание, легкое появление кровоподтеков, раннее развитие одышки, периферические отеки, изменения чувствительности (синдром запястного канала) или ортостатическая гипотензия, следует заподозрить амилоидоз. Для наследственного амилоидоза характерен отягощенный семейный анамнез «нейромышечного» поражения неизвестной этиологии или деменции, для амилоидоза Aβ2M - использование гемодиализа, для амилоидоза АА - наличие хронического воспалительного процесса. Также амилоидоз необходимо исключать у пациентов заболеваниями почек неясного генеза, особенно с нефротическим синдромом, в т.ч. у больных с рестриктивной кардиомиопатией. Амилоидоз более вероятен при наличии обоих упомянутых синдромов. При амилоидозе AA доминирующим органом-мишенью, помимо почек, является печень, поэтому при дифференциальной диагностике причин выраженной гепатомегалии в сочетании с поражением почек следует исключить амилоидоз.
Дополнительная литература :
статья «Сложности диагностики и лечения AL-амилоидоза: обзор литературы и собственные наблюдения» В.В. Рыжко, А.А. Клодзинский, Е.Ю. Варламова, О.М. Соркина, М.С. Сатаева, И.И. Калинина, М.Ж. Алексанян; Гематологический научный центр РАМН, Москва (журнал «Клиническая онкогематология» №1, 2009) [
Амилоидоз - сложное в диагностике и лечении заболевание, при котором довольно часто дают неблагоприятный прогноз. Существует много различных типов болезни, но наиболее известными являются первичный и вторичный амилоидоз. При определении патологии на ранней стадии развития возможен положительный ответ на терапию.
Амилоидоз - расстройство белкового обмена, которое сопровождается отложением в различных тканях и органах специфического белка, называемого амилоидом. Клинические проявления зависят от типа заболевания и в основном определяются как переменные.
Группа системных нарушений под названием “амилоидоз” включает порядка 30 различных типов, отличающиеся между собой спецификой белковых расстройств. Четыре наиболее известных - AL-амилоидоз, AA-амилоидоз, AF-амилоидоз, AH-амилоидоз.
Диагноз может быть поставлен при определении белка в моче или наличии нарушений со стороны внутренних органов без какой-либо причины. Подтверждается заболевание биопсией тканей. Терапия в основном направлена на уменьшение концентрации аномального белка или причину, вызвавшую заболевание.
Видео: Что такое амилоидоз, чем он опасен, как с ним бороться?
Описание
Амилоидоз - системное расстройство, которое подразделяется на несколько типов, классифицируемых как первичные, вторичные или семейные (наследственные).
- Первичный амилоидоз (AL ) является наиболее распространенным типом системного амилоидоза. AL является результатом аномалии (дискразии) плазматических клеток (типа лейкоцитов) в костном мозге и тесно связана с множественной миеломой.
- Вторичный (АА) амилоидоз в своем развитии основывается на определении воспалительного белкового сывороточного амилоида. Нередко сочетается с хроническим воспалительным заболеванием, таким как ревматические заболевания, семейная средиземноморская лихорадка, хроническое воспалительное заболевание кишечника, туберкулез или эмпиема.
- Семейный амилоидоз - это редкий тип амилоидоза, вызванный аномальным геном. Существует несколько аномальных генов, которые могут вызывать развитие патологии, но наиболее распространенный тип наследственного амилоидоза называется АТТР, вызываемый мутациями в транстиретине (ТТР).
- Сенильный амилоидоз , в котором аномальный белок получен из динатного (нормального) транстиретина, является медленно прогрессирующим заболеванием, поражающим сердечную мышцу у пожилых людей.
Амилоидные отложения могут иногда возникать изолированно без признаков системного заболевания. Например, сюда относится единичное поражение мочевого пузыря или амилоидоз трахеи - наиболее распространенные типы изолированного амилоидоза.
Связанный с диализом бета2-микроглобулин амилоидоз - это тип системного амилоидоза, возникающий у людей, которым длительное время проводился удаление накопленных токсинов или отходов из крови путем механической фильтрации. Эта форма амилоидоза, также известная как ABM2 (амилоид, связанная с белком бета-2m), возникает из-за агрегации бета2-микроглобулина, типа амилоидного белка, который очищается в нормально функционирующей почке. Связанный с диализом бета2-микроглобулиновый амилоидоз также встречается у пациентов с почечной недостаточностью в конце течения болезни. При этом болезнь не проявляется у людей с нормальной или умеренно сниженной функцией почек или пациентов после почечной трансплантации.
Признаки
Амилоидоз обычно представляет собой многосистемное заболевание, приводящее к широкому спектру клинических проявлений. Следовательно, пациент может наблюдаться у нескольких специалистов, чаще всего у нефролога, кардиолога или невролога. У большинства пациентов наблюдается поражение нескольких органов, поэтому нередко обнаруживается комбинация признаков, представленных ниже, и при их появлении должно возникнуть подозрение на амилоидоз:
- Почки - чаще всего поражаются при AL, AA и некоторых редких наследственных формах амилоидоза, но редко возникает при семейных формах, вызванных мутациями транстиретина. Чрезмерное количество белка в моче (протеинурия) является обычным проявлением поражения почек и нередко протекает тяжело, что приводит к нефротическому синдрому. Амилоид вызывает избыток мочевины и других азотистых отходов в крови (прогрессирующая азотемия) и является начальным проявлением почечной болезни. Аномальное накопление жидкости (отек), особенно ног и живота, при отсутствии сердечной недостаточности является признаком нефротического синдрома. Также наличие избыточного холестерина в крови (гиперхолестеринемия) может доходить до глубокой степени выраженности. Почки при амилоидозе уменьшаются в размерах, бледнеют и становятся более тяжелыми, но при амилоидозе обычно наблюдаются большие почки. Дополнительно может определяться высокое кровяное давление (гипертония) и тромбоз почечной ткани. Амилоид может накапливаться в других частях мочеполовой системы, например в мочевом пузыре или мочеточниках.
Видео: Елена Малышева. Амилоидоз почек
- Сердце
. При амилоидозе часто поражается сердечная ткань. Амилоидная инфильтрация сердца приводит к утолщению стенки желудочков и развитию сердечной недостаточности. Быстро прогрессирующая застойная сердечная недостаточность с толстыми стенками желудочков представляет собой классическую картину амилоидоза сердца. Миокард неизменно поражается при старческом амилоидозе, ТТР-амилоидозе и почти никогда не участвует во вторичном амилоидозе. Общие симптомы амилоидоза сердца включают:
- увеличенное сердце (кардиомегалия);
- нерегулярное сердцебиение (аритмии);
- шумы в сердце;
- аномалии сердца, наблюдаемые на электрокардиографии (например, низкий вольтаж зубцов).
Застойная сердечная недостаточность является наиболее распространенным осложнением амилоидоза. Узелковые отложения амилоида могут присутствовать на оболочке, которая окружает сердце (перикард), и на внутреннем слое камер сердца или его клапанов (эндокард).
- Нервная система
. Хотя нейропатия менее распространена, чем почечная или сердечная недостаточность, она может представлять значительную проблему при амилоидозе. Довольно часто встречается при амилоидозе AL. Невропатия часто бывает безболезненной и сенсомоторной по своей природе, хотя нейропатическая боль может быть иногда значительной. Из симптомов может определяться:
- сенсорная нейропатия с онемением и покалыванием в ногах, которая прогрессирует по ногам и в конечном итоге переходит на верхние конечности;
- двигательная нейропатия с потерей движения, начинающаяся в ногах и распространяющаяся вверх.
Синдром кистевого туннеля обычно наблюдается не из-за прямого участия нерва, а, скорее, при инфильтрации мягких тканей, что способствует сжатию нерва. При семейном амилоидозе периферическая нейропатия часто сопровождается вегетативной нейропатией, характеризующейся диареей и уменьшением количества пота (гипогидроз), внезапным снижением артериального давления, когда пациент встает (постуральная гипотония), а у мужчин - эректильной дисфункции.
Постуральная гипотензия может быть глубокой и приводить к повторным обморочным (синкопальным) эпизодам. Системный амилоидоз не связан с центральной нервной системой и с болезнью Альцгеймера.
- Органы пищеварения
. Амилоидоз может влиять на печень и селезенку. Поражение последнего органа увеличивает риск его травматического разрыва. Поражение печени распространено при амилоидозе AL. Также бывает встречается при амилоидозе АА, но не наблюдается при семейном амилоидозе ТТР. В большинстве случаев причастность печени является бессимптомной. Наиболее заметными признаками являются
- увеличенная печень (гепатомегалия);
- увеличенная селезенка (спленомегалия).
Как правило, поражение амилоидом печени сопровождается повышение ферментов (особенно щелочной фосфатазы) и других функций органа, нередко обнаруживаемых на ранней стадии. Как правило, функция печени не оказывает существенного влияния на течение болезни на поздних стадиях. Повышение билирубина является неблагоприятным признаком и может предвещать печеночную недостаточность.
Амилоидное накопление в желудочно-кишечном тракте может привести к отсутствию движения (подвижности) пищевода, а также тонкой и толстой кишки. Также могут наблюдаться:
- мальабсорбция;
- изъязвление;
- кровотечение;
- слабая желудочная активность;
- псевдообструкция желудочно-кишечного тракта;
- потеря белка;
- диарея.
- Кожа часто поражается при первичном амилоидозе. Периорбитальная пурпура является результатом хрупкости капилляров и может появляться после кашля, чихания или напряжения при движении кишечника. Нередко пурпурные поражения могут возникать после таких простых действий, как трение век. Инфильтрация мягких тканей вызывает макроглоссию и охриплость голоса, хотя исследование голосовых связок может не выявить нарушения. Повреждения кожи иногда хорошо заметны или настолько незначительны, что для их диагностики требуется использование микроскопа.
Восковидные папулезные поражения могут появляться на лице и на шее. Они также нередко встречаются под мышками (подмышечной областью), вблизи ануса и в паху. Другие области, которые могут быть затронуты, - это слизистые области, ушной канал и язык. Также может присутствовать:
- припухлость;
- кровоизлияния под кожей (пурпура);
- выпадение волос (алопеция);
- воспаление языка (глоссит);
- сухость во рту (ксеростомия).
- Дыхательная система. Проблемы с респираторной системой, которые связаны с амилоидозом, часто развиваются параллельно со сердечными нарушениями. При локализованной форме амилоидоза дыхательные пути могут быть заблокированы амилоидными отложениями в носовых пазухах, гортани, трахее и бронхиальном дереве. Сбор жидкости в плевральном пространстве (плевральный выпот) довольно часто встречается у пациентов с застойной сердечной недостаточностью из-за амилоидоза. Большие рецидивные плевральные выпоты, непропорциональные степени сердечной недостаточности, указывают на плевральный амилоидоз.
Артропатии возникают при амилоидозе из-за накопления амилоидных отложений в синовиальных мембранах. Это происходит при амилоидозе AL и иногда при диализном амилоидозе. Также в патологический процесс могут вовлекаться суставные хрящи или синовиальная мембрана и жидкость. Симптомы сходны с симптомами ревматоидного артрита.
Амилоидные отложения в мышечной ткани могут вызывать слабость в мышцах и мышечные изменения (псевдомиопатии). Симптомы амилоидоза также нередко проявляться при кровотечениях. Это может быть результатом дефицита определенных факторов свертывания крови или небольших амилоидных отложений в кровеносных сосудах внутри кожи.
Причины
Амилоидоз вызван аномальными белками, что способствует образованию фибрилл в одном или нескольких органах, системах или мягких тканях. Эти скопления белка называются амилоидными отложениями, которые способны вызывать прогрессирующее нарушение и полную дисфункцию пострадавшего органа. Обычно белки разрушаются примерно с той же скоростью, что и производятся, но необычно стабильные амилоидные отложения осаждаются быстрее, чем происходит их разрушение.
Причиной первичного амилоидоза (AL) обычно является дисклазия плазматических клеток, приобретенная аномалия плазматических клеток в костном мозге с образованием аномального белка. Обычно образуется избыточное количество белка, накапливаемый в тканях организма в виде амилоидных отложений.
Вторичный амилоидоз (АА ) вызван воспалительным процессом, который является частью основного заболевания. Примерно 50% людей со вторичным амилоидозом имеют ревматоидный артрит в качестве основного заболевания.
Семейный амилоидоз вызывается аномалиями в гене для одного из нескольких конкретных белков. Наиболее распространенная форма наследственного амилоидоза вызвана аномалией (мутацией) в гене для транстиретина. Сообщалось о более чем 100 различных мутациях в транстиретине, и наиболее распространенная мутация была названа V30M. Мутации TTR в основном связаны с амилоидозом, который поражает различные системы органов. Редко мутации в генах белков, которые вызывают амилоидоз, представляют собой афильную цепь фибриногена А, аполипопротеин А1 и А2, гельсолин и цистатин С.
Все наследственные амилоидозы связаны с аутосомно-доминантным типом наследования. Большинство генетических заболеваний определяются по статусу двух копий гена, полученного от отца, и одного от матери. Доминантные генетические расстройства возникают, когда требуется только одна копия аномального гена, чтобы вызвать конкретное заболевание. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека.
Риск передачи аномального гена от пострадавшего родителя к потомству составляет 50% при каждой беременности. Риск одинаковый для мужчин и женщин. Однако не каждый человек, получающий ген, в конечном итоге способен заболеть амилоидозом.
Точная причина амилоидоза бета2-микроглобулина, связанного с диализом, не полностью понятна. Нормально функционирующая почка может очиститься от амилоидного белка, бета2-микроглобулина. У некоторых пациентов с длительным диализом или при непрерывном амбулаторном перитонеальном диализе неспособность почек нормально функционировать приводит к аномальному удержанию и накоплению белка бета2-микроглобулина. У некоторых людей с почечной недостаточностью в конце стадии также развивается эта форма амилоидоза.
Диагностика
Диагноз амилоидоза подозревается после детального изучения истории болезни и клинической картины, но требуется проведение биопсии мышц, костей или жировой ткани для подтверждения наличия амилоида.
Если болезнь подозревается по клиническим признакам, биопсия вовлеченного органа даст самый достоверный результат. Биопсийный материал исследуют микроскопически и окрашивают красителем, называемым конго-красным. Когда диагноз амилоидоза диагностируется при биопсии ткани, проводится обширное обследование больного, что позволяет определить, какие органы затронуты.
Как только с помощью биопсии ткани определен амилоид, необходимо установить тип заболевания. При амилоидозе AL проявляется дискразия плазматических клеток обнаруживаемых в 98% случаев. В 2% случаев В-клеточная лимфома идентифицируется как причина AL.
Конкретные тесты, которые используются для постановки диагноза типа амилоидоза AL, следующие:
- белковый электрофорез крови и мочи;
- биопсия костного мозга с иммунохимическим окрашиванием плазматических клеток;
- бесклеточный анализ легкой цепи.
Диагноз AL амилоидоза подтверждается наличием периорбитальной пурпуры, которая является результатом хрупкости капилляров, или макроглоссии (увеличенный язык).

Диагноз наследственного амилоидоза TTR можно подтвердить, выполнив молекулярно-генетическое тестирование , которое определяет мутации в гене TTR по образцу крови. При отсутствии мутаций транстиретина могут присутствовать очень редкие формы семейного амилоида.
Если пациент является пожилым человеком с клинически изолированной сердечной недостаточностью, наиболее вероятным диагнозом является старческий системный амилоидоз. Это состояние, при котором дистрофический (нормальный) транстиретин осаждается в сердце.
Специфическое иммуноокрашивани е (например, иммуноглоточная электронная микроскопия) доступно в специализированных центрах и является высоко специфичным тестом для определения типа амилоида.
В сложных диагностических случаях масс-спектрометрия способна точно определять молекулярную структуру амилоидных отложений - эта техника используется все чаще.
Метод, называемый сканированием радиоактивной меченой сыворотки амилоида Р , доступен в нескольких центрах Европы, которые специализируются на амилоидозе. Этот тест используется для контроля накопления амилоидных отложений.
У лиц с продолжительным диализом или с почечной недостаточностью на конечной стадии могут проводиться лабораторные тесты, которые позволят проанализировать образцы крови или мочи для выявления повышенного уровня белка B2M.
Стандартная терапия
Стратегия лечения зависит от типа амилоидоза и клинического состояния больного. При амилоидозе AL причиной является аномальная лейкоцитарная клетка (как правило, плазменная клетка), и поэтому в основу терапии этого типа амилоидоза входит химиотерапия, направленная на искоренение этих клеток.
На протяжении многих лет использовались мелфалан и дексаметазон с пероральным или внутривенным путем введения, нередко сочетающиеся с аутологичной поддержкой стволовых клеток.
Оба препарата одинаково эффективны, но лечение и побочные эффекты различны. Высокая доза мелфалана с поддержкой стволовых клеток - это курсовое лечение, которое часто включает 2-3-недельное пребывание в больнице и несколько месяцев восстановления. Использование пероральной формы мелфалана ежемесячными курсами менее токсично, но связано с более высоким риском развития лейкемии.
Более новые препараты, активные в отношении множественной миеломы (другое заболевание аномальных плазматических клеток), такие как бортезомиб или леналидомид, также очень эффективны в отношении AL и, как было доказано, дают некоторое преимущество пациентам с рецидивирующим заболеванием. Часто эти препараты включаются в предварительное лечение.
В настоящее время большинство пациентов, не используемых мелфалан с высокой дозой с поддержкой стволовых клеток, получают авангардную терапию. Комбинация бортезомиба, циклофосфамида и дексаметазона связана с хорошей переносимостью и быстрыми ответами. Лечение амилоидоза для любого больного должно быть составлено персонально с учетом особенностей ситуации.
Два наиболее важных фактора долговременной выживаемости с AL - это наличие / степень поражения сердца и гематологический ответ на терапию.
Есть несколько новых лекарств, предназначенных для стимуляции резорбции амилоида из пораженных органов. Их применение может обеспечить способность лечить больные органы напрямую. Наиболее продвинутый из этих исследований - с NEOD001, который показал некоторую пользу для пациентов, чья основная болезнь клеток плазмы уже лечилась. В настоящее время метод изучается в сочетании с терапией на основе бортезомиба в начальной стадии.
Поддерживающая терапи я (лечение застойной сердечной недостаточности, внимание к питанию, лечение вегетативной нейропатии и т. д.) является очень важным элементом лекарственного воздействия. Учитывая сложность заболевания, рекомендуется, чтобы лечение проводилось в специализированном центре по амилоидозу или, по крайней мере, больной должен пройти первоначальную оценку в таком медицинском учреждении с продолжением лечения по месту жительства.
Семейный амилоидоз устраняется, если это возможно, путем удаления первопричины аномальной ТТР-продукции. Поскольку доминирующим источником является печень, трансплантация органа в настоящее время является предпочтительным выбором у тщательно отобранных пациентов, болезнь которых находится на допустимых стадиях развития. Тафамидис - препарат, недавно одобренный для терапии семейной амилоидной полинейропатии. Это лекарство тестируется в текущих испытаниях для других форм заболевания. Патисиран и ревусиран также тестируют в отношении воздействия на ATTR форму амилоидоза, при этом ориентация направлена на снижение уровней TTR, который образует амилоид.