Фармацевтическая эквивалентность. Актуальная тема эквивалентность воспроизведенных лекарственных средств: фармацевтические аспекты. Причины неполной биоэквивалентности
Фармацевтическая эквивалентность
Лекарственные препараты фармацевтически эквивалентны, если они содержат одни и те же активные субстанции в одинаковом количестве и в одинаковой лекарственной форме, отвечают требованиям одних и тех же или сходных стандартов и идентичны по силе действия или концентрации активных веществ. Часто, несмотря на одинаковое содержание действующего вещества, дженерический препарат отличается от оригинального по составу вспомогательных веществ
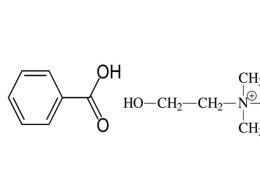
Состав оригинального препарата Вигамокс и дженерического Моксицина в пересчете на 5 мл раствора
- · Вигамокс (28)
- · Моксицин (29)
Действующее веществом оксифлоксацина гидрохлорид 0,02725 г моксифлоксацина гидрохлорид 0, 02725 г
Консервант бензалкония хлорид
Другие вспомогательные вещества натрия хлорид натрия хлорид
кислота борная
кислота хлороводородная и/или натрия гидроксид (для регулировки рН)
вода для инъекций
В состав дженерического моксифлоксацина гидрохлорида входит консервант, оригинальный препарат Вигамокс консерванта не содержит.
Биоэквивалентность
Два лекарственных препарата считаются биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и после назначения в одинаковой дозе являются сходными, обеспечивая должную эффективность и безопасность. Под биодоступностью понимается скорость и доля всасывания активного ингредиента или активного компонента лекарства, которое начинает действовать в точке приложения.
В сущности, биоэквивалентность -- это эквивалентность скорости и степени всасывания оригинала и дженерика в одинаковых дозах по концентрации в жидкостях и тканях организма. Достоверность результатов сравнительного исследования биоэквивалентности во многом зависит от соблюдения требований (GМP -- надлежащая клиническая практика) и должно быть независимым, многоцентровым, рандомизированным, контролируемым, длительным.
Если дженерик разрешен к применению в других странах, он регистрируется в РФ по упрощенной схеме (без определения биоэквивалентности). Таким образом, при регистрации зарубежных дженериков в РФ мы в значительной степени доверяем досье, представляемым фармацевтическими компаниями. Такая «доверчивость» в ряде случаев дорого обходится пациентам, т.к. дженерики по своим фармакокинетическим свойствам могут не соответствовать оригинальному препарату. На примере контрольной проверки биоэквивалентности дженериков оригинальному кларитромицину C.N. Nightingale и соавторы сравнили оригинальный препарат кларитромицина с 40 копиями в отношении биоэквивалентности, применив стандарты американской фармакопеи. Исследование показало, что 70% дженериков растворяются значительно медленнее оригинального препарата, что критично для их усвоения. 80% дженериков отличаются от оригинала по количеству действующего начала в одной единице продукта. Количество примесей, не имеющих отношения к действующему началу, в большинстве образцов больше, чем в оригинале. В «лучшем» дженерике их было 2%, в «худшем» -- 32%. Наличие примесей определило выраженность побочных реакций.
С аналогичной ситуацией сталкиваются и офтальмологи. Congdon N.G. и соавторы (2001) по результатам рандомизированного двойного слепого исследования установили преобладание случаев раздражения конъюнктивы и роговицы в связи с местным применением дженерического НПВС -- диклофенака по сравнению с больными, получавшими брендовый препарат.
Вопросы взаимозаменяемости лекарственных препаратов - это наиболее конфликтные и сложные вопросы фармацевтического рынка. Взаимоотношения между оригинальными и воспроизведенными препаратами (или дженериками) далеко не безоблачны.Особенности патентной защиты оригинальных препаратов
Действительно, фирму-разработчика оригинального препарата можно понять. Колоссальные средства, которые тратятся на поиск молекулы лекарственного вещества, исследование лекарства, выведение его на рынок, тщательный мониторинг возможных неблагоприятных эффектов и взаимодействий, через несколько лет, когда продолжает действовать патентная защита, оказываются, казалось бы, безвозвратно потерянными.
Дженерический препарат, который начинают воспроизводить зачастую несколько фирм одновременно, «наследует» все те свойства, те затраты сил, времени и средств, что и препарат оригинальный. И можно сколь угодно утверждать, что оригинал всегда остается оригиналом, а воспроизведенное средство - всего лишь воспроизведенным средством. Общее международное непатентованное название делает эти препараты аналогичными для потребителя, а генерик, из-за, как правило, меньшей цены, более привлекательным.
Производители оригинальных фармацевтических брендов защищают свои исключительные права различными путями, в первую очередь с помощью патентного права. Реализация патентной защиты на ту или иную молекулу, лежащую в основе лекарственного вещества, предусматривает запрет ее воспроизводства на срок, длительность которого различается в разных странах, но в среднем она
Препараты дженерики и копии
Препаратом-дженериком называется лекарственный препарат, срок действия патентной защиты на который уже закончился. Соответственно, препарат-генерик не является исключительной собственностью фармацевтической компании, которая его разработала или владела первой лицензией на его продажу.
Копии - это лекарственные препараты, которые представлены на рынках стран со слабой или отсутствующей патентной защитой химических молекул - активных ингредиентов лекарственных средств.
В сущности, отличием копированного препарата от препарата-дженерика является только нарушение юридических правил воспроизводства лекарственного препарата (ущемление прав патентообладателя).
В конечном итоге, в странах с развитой патентной защитой потребители сталкиваются с оригинальным препаратом, и лишь затем препаратам генерической линии приходится завоевывать свое место на рынке.
Во-вторых, традиции советской медицины и длительное присутствие на рынке исключительно отечественных препаратов или препаратов, произведенных в бывших странах СЭВ, спровоцировали некоторое смещение в восприятии брендовых названий. Так, Пирацетам для российских врачей - в первую очередь воспроизведенный препарат Ноотропил; Ко-тримоксазол более известен под названием Бисептол; Ренитек (эналаприла малеат) вошел в обиход под наименованием его наиболее успешного на российском рынке генерика - Энапа; оригинальный ципрофлоксацин (Ципробай) подменяется названиями Цифран и Ципролет.
Таким образом, специфика рынка диктует восприятие оригинальных названий, что определяет субъективный выбор между оригиналом и генериком в пользу последнего.
В-третьих, как и любая страна с высоким уровнем государственного протекционизма в области медицины, Россия выбирает дженерические препараты, потому что они дорогостоящие. Это и определяет заполнение генериками наиболее массового сектора медицины - медицины условно бесплатной.
Активная антидженерическая политика, проводимая разработчиками оригинальных лекарств, привела к тому, что сам термин генерик приобрел некую оскорбительность. Это способствует тому, что имплицидными характеристиками препарата-генерика становятся его второсортность, недостаточная изученность, неуточненный профиль безопасности. Между тем, для этого нет никаких объективных оснований.
При оценке генерических препаратов следует иметь ввиду следующее.
- Дженерик содержит то же активное лекарственное вещество (субстанцию), что и оригинальный (патентованный) препарат.
- Дженерик отличается от оригинального препарата вспомогательными веществами (неактивными ингредиентами, наполнителями, консервантами, красителями и др.).
- Различия наблюдаются и в самом технологическом процессе производства дженериков.
Зачастую термин «дженерик» неверно заменяется термином «эквивалентное лекарственное вещество». Собственно, подобный термин является бессмыссленным, так как не существует понятия «эквивалентность лекарственных веществ». Выделяются следующие виды эквивалентности: фармацевтическая, биологическая и терапевтическая. В странах Евросоюза и в США используют дефиниции фармацевтической эквивалентности лекарственных веществ.
1. Взаимозаменяемость лекарственных препаратов для медицинского применения определяется в порядке , установленном Правительством Российской Федерации, на основании следующих параметров:
1) эквивалентность (для биоаналоговых (биоподобных) лекарственных препаратов (биоаналогов) - сопоставимость) качественных и количественных характеристик фармацевтических субстанций (использование различных солей, эфиров, комплексов, изомеров, кристаллических форм и других производных одного и того же действующего вещества не является препятствием для взаимозаменяемости лекарственных препаратов, если при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения);
2) эквивалентность лекарственной формы (под эквивалентными лекарственными формами понимаются разные лекарственные формы, имеющие одинаковые способ введения и способ применения, обладающие сопоставимыми фармакокинетическими характеристиками и фармакологическим действием и обеспечивающие также достижение необходимого клинического эффекта. Различия лекарственных форм не являются препятствием для их взаимозаменяемости, если при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения);
3) эквивалентность или сопоставимость состава вспомогательных веществ лекарственного препарата для медицинского применения (различия состава вспомогательных веществ лекарственного препарата для медицинского применения не являются препятствием для их взаимозаменяемости, если при проведении исследования биоэквивалентности лекарственного препарата для медицинского применения или в случае невозможности проведения этого исследования при проведении исследования терапевтической эквивалентности лекарственного препарата для медицинского применения доказано отсутствие клинически значимых различий фармакокинетики и (или) безопасности и эффективности лекарственного препарата для медицинского применения. При этом различия состава вспомогательных веществ не должны приводить к риску возникновения серьезных нежелательных реакций у отдельных групп пациентов или повышения частоты их возникновения);
4) идентичность способа введения и применения;
5) отсутствие клинически значимых различий при проведении исследования биоэквивалентности лекарственного препарата или в случае невозможности проведения этого исследования отсутствие клинически значимых различий показателей безопасности и эффективности лекарственного препарата при проведении исследования терапевтической эквивалентности. Данный параметр не применяется в отношении воспроизведенных лекарственных препаратов, указанных в части 10 статьи 18 настоящего Федерального закона. В отношении биоаналоговых (биоподобных) лекарственных препаратов (биоаналогов) данные об отсутствии клинически значимых различий безопасности, эффективности и иммуногенности лекарственного препарата по результатам проведения клинических исследований предоставляются в порядке, установленном настоящей частью;
6) соответствие производителя лекарственного средства требованиям надлежащей производственной практики.
2. Сравнение параметров регистрируемых лекарственных препаратов для медицинского применения осуществляется комиссией экспертов экспертного учреждения при проведении экспертизы таких лекарственных препаратов в процессе их государственной регистрации. Выводы экспертов о взаимозаменяемости или невзаимозаменяемости лекарственных препаратов для медицинского применения, сделанные в результате этого сравнения, оформляются в виде приложения к заключению экспертов по форме , утвержденной уполномоченным федеральным органом исполнительной власти.
3. Положения настоящей статьи не распространяются на референтные лекарственные препараты, лекарственные растительные препараты, гомеопатические лекарственные препараты и лекарственные препараты, которые разрешены для медицинского применения в Российской Федерации более двадцати лет и в отношении которых невозможно проведение исследования их биоэквивалентности.
Конечной целью лекарственной политики в любой стране мира является обеспечение населения безопасными, эффективными, качественными и доступными медикаментозными средствами. Один из ключевых моментов в этой политике – широкое использование генерических лекарств.
Ю.С. Рудык, Институт терапии имени Л.Т. Малой АМН Украины, г. Харьков
 Наиболее часто генерики применяются при социально значимых заболеваниях, имеющих высокую распространенность (артериальной гипертонии, хронической сердечной недостаточности, туберкулезе, сахарном диабете и др.). В связи с этим очевидно, что благоприятного влияния на течение и исход социально значимых заболеваний можно добиться только при использовании относительно доступных и высококачественных генериков .
Наиболее часто генерики применяются при социально значимых заболеваниях, имеющих высокую распространенность (артериальной гипертонии, хронической сердечной недостаточности, туберкулезе, сахарном диабете и др.). В связи с этим очевидно, что благоприятного влияния на течение и исход социально значимых заболеваний можно добиться только при использовании относительно доступных и высококачественных генериков .
Согласно определению ВОЗ, под термином «генерик» понимают лекарственный препарат, используемый в медицинской практике взаимозаменяемо с инновационным (оригинальным) средством, производящийся, как правило, без лицензии от компании-создателя и реализуемый после истечения срока действия патента или других исключительных прав.
Генерический препарат должен удовлетворять следующим критериям :
- содержать такой же активный ингредиент, как и оригинальный препарат;
- иметь аналогичную биодоступность;
- выпускаться в той же лекарственной форме;
- сохранять качество, эффективность и безопасность;
- не иметь патентной защиты;
- иметь более низкую стоимость по сравнению с оригинальным препаратом;
- соответствовать фармакопейным требованиям, производиться в условиях GMP (надлежащей производственной практики);
- иметь те же показания к применению и меры предосторожности.
Как показывает клиническая практика, лекарственные препараты, содержащие одинаковые активные ингредиенты в тех же фармацевтических формах и дозах, но производящиеся на различных предприятиях, могут существенно отличаться как по терапевтической эффективности, так и по частоте возникновения побочных реакций, предусмотренных в инструкциях по их медицинскому применению.
В директиве ЕС 2001/83 дается также определение по существу аналогичных препаратов. Лекарственный препарат является по существу аналогичным оригинальному препарату, если он удовлетворяет критериям одного и того же количественного и качественного состава относительно действующих веществ, одной и той же лекарственной формы и является биоэквивалентным, если только с научной точки зрения не очевидно, что он отличается от оригинального препарата относительно безопасности и эффективности.
Одним из главных вопросов как для врача, так и для пациента является проблема взаимозаменяемости генерического и оригинального препаратов .
Международное сообщество, национальные службы здравоохранения заинтересованы в разработке и внедрении в практику научно обоснованных критериев оценки эффективности и безопасности генерических препаратов, которые производятся различными фирмами.
Согласно современным представлениям, соответствие генерика и препарата-бренда основывается на трех важнейших компонентах, обозначаемых как фармацевтическая, фармакокинетическая и терапевтическая эквивалентность .
В странах Европы считается, что лекарственные препараты являются фармацевтически эквивалентными , если они содержат аналогичные активные субстанции в одинаковом количестве и в одинаковой лекарственной форме, отвечают требованиям одних и тех же или сходных стандартов .
По американскому определению, фармацевтически эквивалентные лекарственные препараты содержат одинаковые активные ингредиенты в одинаковой лекарственной форме, предназначены для одного способа введения и идентичны по силе действия или концентрации активных веществ .
Но фармацевтически эквивалентные средства не обязательно будут являться терапевтически эквивалентными, т.е. такими, после применения которых в одинаковой молярной дозе воздействие с точки зрения эффективности и безопасности фактически одинаково. Так, зарегистрированный в Российской Федерации эритромицин при внутривенном введении с высокой частотой вызывал тромботические осложнения, тогда как в странах Европы эритромицин компании Abbott широко используется для внутривенного введения, считаясь при этом наиболее безопасным антибиотиком-макролидом для внутривенных инфузий .
Важную роль в безопасности применения лекарственных средств играют вспомогательные вещества. При создании генерических препаратов следует требовать сохранения оригинального состава вспомогательных вещества, который, однако, не всегда известен. Использование вспомогательных ингредиентов в препаратах-генериках регламентируется на основе рекомендаций ВОЗ .
При оценке фармакокинетической эквивалентности (или биоэквивалентности) сопоставляются особенности всасывания и распределения лекарств в организме человека. Согласно определению ВОЗ, «два лекарственных препарата считаются биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и при назначении в одинаковой дозе обеспечивают должную эффективность и безопасность».
Определения, принятые в странах Европейского Союза (ЕС) и в США, несколько отличаются.
Согласно европейской формулировке, два лекарственных препарата биоэквивалентны, если они фармацевтически эквивалентны или альтернативны и если их биодоступность (скорость и степень всасывания) после введения в одинаковой молярной дозе сходна в такой степени, что их эффективность и безопасность в основном одинаковы .
В соответствии с американским определением, биоэквивалентные лекарственные препараты – это фармацевтически эквивалентные или фармацевтически альтернативные средства, которые имеют сравнимую биодоступность при исследовании в сходных экспериментальных условиях .
Исследование биоэквивалентности, по сути, представляет (для перорально применяемых препаратов) испытание сравнительной биодоступности. Для каждого исследуемого лекарства должны быть определены основные фармакокинетические параметры, характеризующие полноту абсорбции: площадь под кривой «концентрация–время» (AUC), скорость абсорбции (C max , T max) и скорость выведения активного вещества (K el , T 1/2). Для заключения об отсутствии различий в этих параметрах применяется дисперсионный анализ и выполняется расчет 90% доверительных интервалов. Для подтверждения эквивалентности требуется, чтобы 90% доверительные интервалы соотношений параметров биодоступности исследуемого препарата не выходили за пределы -80 и +125% показателей референтного препарата.
При этом важно заметить, что нельзя говорить о биоэквивалентности препаратов, если неизвестно наверняка, где и как произведено лекарственное средство. Если нет уверенности в том, что производственный участок, где выпускается этот препарат, соответствует требованиям GMP, бессмысленно заниматься изучением биоэквивалентности, как и другими клиническими испытаниями, потому что качество препаратов не сохраняется от серии к серии. В глобальном смысле GMP – это пошаговое, планомерно-поэтапное «встраивание» качества в препарат. В этом плане исследование биоэквивалентности – лишь часть общей системы обеспечения качества лекарственных средств.
Все генерики должны иметь доказанную биоэквивалентность, поскольку теоретически только биоэквивалентные лекарственные препараты могут обладать сходными клинической эффективностью и профилем безопасности.
В 1984 г. президентом США был подписан закон, требовавший от FDA (Управление по контролю за пищевыми продуктами и лекарственными средствами) сделать общедоступным перечень одобренных рецептурных и безрецептурных лекарственных средств. Этим законом впервые было введено новое допущение, что биоэквивалентные препараты являются терапевтически эквивалентными и поэтому взаимозаменяемыми. Издание «Получившие разрешение на маркетинг лекарственные средства с оценками терапевтической эквивалентности» (Approved Drug Products with Therapeutic Equivalence Evaluations) – список, который принято называть «Оранжевой книгой» (Orange Book) , – идентифицирует препараты, одобренные FDA на основании их безопасности и эффективности. Говоря о статусе «Оранжевой книги», необходимо отметить, что информируя о своей оценке терапевтической эквивалентности препаратов при помощи списка, FDA предлагает свои рекомендации общественности, специалистам и уполномоченным органам по выбору лекарственного средства. Такую оценку не нужно рассматривать как запрет использовать тот или иной препарат либо как свидетельство того, что один их них предпочтительнее другого. «Оранжевая книга» служит большей частью не для различения многоисточниковых лекарственных средств между собой, а информирует о том, удалось ли с помощью доступных инструментов решить проблему доказательства их терапевтической эквивалентности референтному препарату или нет. Терапевтическая эквивалентность является научным суждением, тогда как практика генерической замены, имеющая целью экономию средств, основывается также на социальных и экономических аспектах.
Появление «Оранжевой книги» связано с тем, что в целях экономии средств в системе здравоохранения практически во всех штатах США были приняты законы и/или нормативные акты, стимулирующие выполнение генерической замены. Претворение в жизнь этих законов требовало создания позитивного или негативного перечня лекарств (тех, которыми либо можно, либо нельзя заменять оригинальный препарат). Специалисты FDA создали единый формуляр лекарственных средств, в котором оценка терапевтической эквивалентности препаратов была представлена в виде буквенного кода. Система буквенных кодов, описывающих терапевтическую эквивалентность, позволяет быстро определить, установлена ли биоэквивалентность определенного препарата референтному (первая буква), и получить дополнительную информацию об оценке FDA (вторая буква). Две основные категории, к которым могут быть отнесены генерические препараты, обозначены буквами А и В. К категории А относят препараты, терапевтически эквивалентные другим фармацевтически эквивалентным продуктам, для которых:
- нет известных или предполагаемых проблем с биоэквивалентностью; они обозначаются буквами AA, AN, AO, AP или AT в зависимости от лекарственной формы;
- действительные или потенциальные проблемы с биоэквивалентностью могут быть разрешены путем адекватных доказательств биоэквивалентности; в таких случаях применяют обозначение АВ.
При помощи кода В обозначают препараты, которые FDA в настоящее время считает неэквивалентными терапевтически другим фармацевтически эквивалентным продуктам, то есть действительные или потенциальные проблемы биоэквивалентности не могут быть разрешены путем адекватного установления биоэквивалентности. Часто проблема заключается в определенной лекарственной форме, а не в действующем веществе. В таких случаях применяют обозначения BC, BD, BE, BN, BP, BR, BS, BT, BX или B.
В свое время FDA опубликовало проект руководства по вопросам деятельности фармацевтических компаний, а также предприятий, которые являлись собственностью или подчинялись влиянию распространителей (так называемых спонсоров) медицинской продукции. Необходимость проведения публичных слушаний и обсуждения проекта была обусловлена тем, что отдельные лица и группы людей обращались в государственные законодательные органы, фармацевтические организации, а также в комитеты по контролю за использованием лекарственных средств, выражая озабоченность по поводу проблемы взаимозаменяемости некоторых лекарств, в частности препаратов с ограниченным терапевтическим индексом. Особенно их интересовало, не изменятся ли показатели безопасности и эффективности таких лекарств, если вместо препарата известной фирмы-производителя начать применять препарат, признанный FDA терапевтически эквивалентным, но не защищенный зарегистрированной торговой маркой. С разъяснениями на эту тему в 1998 г. было опубликовано письмо специального уполномоченного по делам здравоохранения FDA Стюарта Л. Найтингейла. Ниже приводится его резюме : «Основываясь на определении терапевтической эквивалентности препаратов, FDA сделало заявление:
- дополнительные клинические тесты не обязательны при замене препарата известной фирмы средством с незарегистрированной торговой маркой;
- не нужно предпринимать специальные меры предосторожности при изменении формулы или процесса производства препарата при условии, что эти изменения одобрены FDA в соответствии с законами и правилами FDA;
- как указано в «Оранжевой книге», по мнению FDA, можно ожидать, что препараты, признанные терапевтически эквивалентными, будут иметь одинаковый клинический эффект, независимо от того, является ли препарат известным или новым;
- нет необходимости рассматривать любой класс лекарственных средств отлично от другого класса, если FDA определила терапевтическую эквивалентность для препаратов, о которых идет речь».
Согласно FDA, терапевтически эквивалентными считаются препараты, удовлетворяющие следующим общим требованиям:
а) доказаны их эффективность и безопасность;
б) они фармацевтически эквивалентны, а именно:
- содержат одинаковое количество идентичных активных ингредиентов в одинаковой лекарственной форме и предназначены для одного способа введения;
- соответствуют требованиям по силе действия, качеству, чистоте и идентичности;
в) являются биоэквивалентными, а именно:
- нет известных или потенциальных проблем с биоэквивалентностью и они соответствуют стандарту in vitro или
- если имеющиеся известные или потенциальные проблемы могут быть устранены проведением исследований биоэквивалентности;
г) адекватные указания в инструкции;
д) произведены в соответствии с требованиями GMP.
Согласно определению ВОЗ, два лекарственных средства считаются терапевтически эквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность лекарственного вещества и после введения в одинаковой молярной дозе их действие обеспечивает соответствующую эффективность и безопасность .
Таким образом, терапевтическая эквивалентность является основным требованием взаимозаменяемости лекарственных препаратов .
Определение биоэквивалентности лекарственных средств – основной критерий медико-биологического контроля качества генерических препаратов, принятый для стран ЕС , США , Российской Федерации и др.
Считается, что если биоэквивалентность препаратов доказана, нет необходимости проводить дополнительные клинические испытания генерических средств, поскольку наличие биоэквивалентности свидетельствует, что все показатели эффективности и безопасности исследуемого препарата сопоставимы. Испытания с целью установления биоэквивалентности – это клинические исследования, субъектами которых являются здоровые добровольцы или больные, которым показано назначение исследуемого лекарственного средства.
Оценка биоэквивалентности генерических препаратов жестко регламентируется соответствующими международными и национальными стандартами. В настоящее время в Украине в связи с интенсивным расширением фармацевтического рынка возрастает конкуренция среди аналогов лекарственных средств различного производства. Биоэквивалентность многих из них (особенно это касается отечественных лекарственных средств) не доказана. Проведенные клинические испытания по ограниченной программе этих препаратов не всегда могут дать достаточно объективную информацию об их эффективности и безопасности .
В методических рекомендациях ВОЗ по определению взаимозаменяемости аналогичных препаратов, доступных из различных источников (так называемые многоисточниковые лекарственные средства), отмечается, что для подтверждения терапевтической эквивалентности чаще всего используется биоэквивалентность . Вместе с тем возможны и другие подходы , а именно:
- сравнительное определение фармакодинамических характеристик (например, расширение зрачка, изменение сердечного ритма или артериального давления), когда фармакодинамический отклик легче измеряем или более надежен, чем фармакокинетические параметры, или для лекарственных средств местного действия;
- сравнительные клинические испытания в ограниченном объеме, когда ни фармакокинетические, ни фармакодинамические исследования не дают убедительных доказательств;
- испытания in vitro, например определение растворимости дозированной формы (dissolution test), в том числе и в форме профиля растворимости, установленного по нескольким точкам.
Наконец, в ряде случаев специальных доказательств терапевтической эквивалентности не требуется, к примеру при условии, что все химические (например, профиль примесей), фармацевтические (например, стабильность) и производственные (GMP) показатели соответствуют таковым избранного эталона. Иными словами, считается, что в этих случаях соответствие технических параметров само по себе гарантирует терапевтическую эквивалентность. Во всех случаях речь идет о сравнительных испытаниях с препаратами, терапевтическая эффективность которых считается доказанной.
Исходя из вышесказанного очевидно, что терапевтическая эквивалентность включает в себя фармацевтическую эквивалентность и один из критериев :
- изучение биоэквивалентности на людях;
- исследование фармакодинамики на людях;
- клинические испытания;
- тест растворения in vitro (в некоторых случаях).
Производство и контроль качества генериков зависят также от вспомогательных веществ. Требования к ним должны быть такими же, как и к активной субстанции. Любое изменение в составе наполнителей или оболочки лекарства может существенно изменить качество препарата, его биодоступность, привести к токсическим или аллергическим явлениям.
Концепция терапевтической эквивалентности применима лишь к лекарственным средствам, содержащим одинаковые активные ингредиенты, и не распространяется на разные терапевтические препараты, используемые в одинаковых клинических ситуациях (например, парацетамол и ацетилсалициловая кислота, назначаемые при головной боли).
Лекарственное средство, соответствующее вышеуказанным критериям терапевтической эквивалентности, считается таковым, даже если оно отличается по некоторым характеристикам, таким как форма, риска на таблетке, упаковка, эксцепиенты (включая красители, консерванты), срок годности и минимальные отличия в инструкции (например, наличие специфической информации о фармакокинетике), а также условия хранения. Если подобные отличия важны в лечении конкретного пациента, врач может требовать, чтобы ему был отпущен из аптеки конкретный бренд. Не считая этого ограничения, FDA полагает, что препараты, классифицированные как терапевтически эквивалентные, можно заменять, полностью полагаясь на то, что в результате замены сохранятся эффекты и профиль безопасности, ожидаемые от назначенного препарата.
Надо признать, что как в странах ЕС, так и в США многие специалисты ставят под сомнение фармакокинетическую эквивалентность как единственный способ оценки взаимозаменяемости лекарств. В ряде публикаций указывается на существенные методологические недостатки изучения биоэквивалентности лекарственных средств, которые могут привести к тому, что существующие различия между брендовыми и генерическими препаратами не будут выявлены . Согласно европейским требованиям и регламенту FDA, отдельные показатели фармакокинетики могут отличаться до 20%. Считается, что колебания концентрации активного компонента в плазме крови в пределах от -20 до +25% клинически не значимы, однако для пациентов пожилого возраста или других уязвимых групп больных даже такие незначительные изменения концентрации лекарственного вещества могут повышать риск побочных эффектов.
Предполагается, например, что определенные ограничения могут быть связаны с существованием лекарственных средств, характеризующихся сравнительно небольшим разбросом терапевтических концентраций препарата в плазме крови (некоторые антидепрессанты – пароксетин, флуоксетин, циталопрам) и/или нелинейной фармакокинетикой (нормотимики и противоэпилептические препараты) .
В данной ситуации даже небольшие изменения этого параметра, вполне укладывающиеся в допустимые пределы теста биоэквивалентности (от -20 до +25%), могут оказаться значимыми для клинической эффективности и/или переносимости .
Следовательно, возможны значительные расхождения свойств брендовых и генерических лекарственных средств . Например, при показателях биоэквивалентности ниже 100% препарат может оказаться неэффективным. Напротив, при повышении рассматриваемого показателя следует ожидать возрастания числа побочных эффектов. Особые опасения вызывают лекарственные средства с низким терапевтическим индексом (разницей между минимальной эффективной дозой препарата и его максимально токсичной дозой) – дигоксин, фенитоин, карбамазепин, циклоспорин, варфарин. Такая ситуация требует ужесточения и расширения требований к фармакокинетическим исследованиям. Обсуждается вопрос об уменьшении различий в параметрах до 10-15%, что позволит уменьшить число препаратов с пограничными фармакокинетическими параметрами.
Еще одно ограничение накладывает на использование результатов теста биоэквивалентности существование лекарственных средств (сертралин, флуоксетин, хлорпромазин, клозапин) со значительной вариабельностью фармакокинетических показателей, которая зависит, в частности, от сложности процессов метаболизма препарата (система цитохромов, наличие нескольких путей выведения и т.д.). Такая изменчивость может носить «интраиндивидуальный» характер. В одном случае она связана, например, с генетическим полиморфизмом цитохромов, который наблюдается в различных популяциях населения, в другом – с функциональным состоянием этих ферментов, меняющимся у одного и того же человека под влиянием различных внешних факторов (например, употребление грейпфрутового сока). Следовательно, результаты теста биоэквивалентности, выполненного на небольшой группе добровольцев, употреблявших сходную диету, могут оказаться невалидными для реальных клинических условий .
Критически воспринимается и тенденция использовать во время исследования биоэквивалентности единственную суточную дозу препаратов .
Известно, что многие препараты (амиодарон, препараты дигиталиса, психотропные средства) назначаются многократно в течение определенного промежутка времени и для получения клинического эффекта требуется достижение устойчивой (терапевтической) концентрации препарата в плазме крови и/или ткани, которая может быть значительно выше той, что используется при проведении исследований биоэквивалентности на здоровых добровольцах .
Следует также иметь в виду, что в реальной клинической практике воспроизведенные лекарственные средства длительное время принимают пациенты разного возраста, пола, массы тела, часто страдающие коморбидной (сопутствующей) патологией. В такой ситуации фармакокинетические свойства брендовых и генерических лекарственных средств в силу существования между ними даже небольших химических отличий могут существенно различаться. Определенное значение приобретает патология желудочно-кишечного тракта. У пациентов с таким заболеванием достаточно сложный механизм всасывания лекарственного вещества легко нарушается. При этом даже незначительные различия в химическом составе брендового и генерического препаратов могут привести к нарушению их биоэквивалентности .
В частности, может возникнуть ситуация, когда инертные составы (наполнители), используемые в генериках, при назначении в одной-единственной дозе, не влияя на всасывание, распределение и метаболизм препаратов, при длительном применении могут оказывать воздействие на функциональное состояние желудочно-кишечного тракта, печени или почек таким образом, что фармакокинетическая эквивалентность препаратов существенно нарушается .
Как пример можно привести различные составы вспомогательных веществ оригинальных и генерических препаратов ницерголина, широко применяемых пациентами разного возраста, в том числе пожилыми больными, часто страдающими широким спектром сопутствующих заболеваний внутренних органов.
С наличием сопутствующей соматической патологии связана и другая проблема, значительно затрудняющая клиническое использование результатов теста биоэквивалентности. В отличие от здоровых добровольцев пациенты с сопутствующей патологией часто вынуждены принимать различные соматотропные лекарственные средства, в частности усиливающие или ослабляющие перистальтику, влияющие на разрушение лекарственного вещества в кишечнике. Не исключено, что это влияние в силу существующих, хотя и минимальных, отличий химического состава оригинального и генерического препаратов может оказаться неоднозначным. Соответственно возникают условия для изменения биоэквивалентности этих препаратов .
Рассмотренные возражения не являются лишь теоретическими соображениями. В соответствующих публикациях имеется много сведений о результатах перепроверки биоэквивалентности различных препаратов. Эти данные свидетельствуют о том, что значительная часть генериков не выдерживает такого тестирования. Так, проведенный в Великобритании в 1995-1996 гг. анализ 2427 воспроизведенных лекарственных средств обнаружил 228 существенных различий . Не менее поразительные данные получены в США. FDA выявило, что до 20% имеющихся на территории страны брендовых и генерических препаратов не являются биоэквивалентными и, следовательно, не могут быть взаимозаменяемыми .
Приводятся примеры клинической неэквивалентности препаратов эналаприла. Показано, что клиническая эффективность по достижению целевого уровня артериального давления у пациентов с артериальной гипертензией 4 генерических эналаприлов известных производителей была ниже, чем у оригинального препарата (Ренитек, MSD). Исследуемые генерики были фармакокинетически эквивалентны Ренитеку. На основании полученных результатов авторы сделали вывод о неодинаковой терапевтической эквивалентности воспроизведенных препаратов эналаприла .
О терапевтической неэквивалентности оригинального индапамида (Арифон, Servier) и его генериков у пациентов с артериальной гипертензией сообщают В.И. Петров и соавт. , при этом фармакокинетические профили сравниваемых препаратов совпадали .
Особое значение эквивалентность генериков имеет для антимикробных препаратов, так как низкая антимикробная активность может приводить к снижению клинической эффективности терапии, что особенно важно при лечении тяжелых больных, и быстрому распространению резистентных форм микробов. Недавно проведенное исследование микологической активности оригинального флуконазола (Дифлюкан, Pfizer) и генерических препаратов показало, что активность генериков против различных видов грибов рода Candida в 2 раза ниже, чем у Дифлюкана. При этом генерики были биоэквивалентны оригинальному препарату .
В одной из публикаций приводятся данные сравнительного анализа качества оригинального кларитромицина производства компании Abbott и 40 его генериков из 13 стран Азии и Латинской Америки. Оказалось, что в 8 препаратах содержание активного вещества не соответствовало стандартам компании-разработчика, у 28 генериков количество высвобождающегося при растворении активного компонента было значительно ниже, чем у оригинального, хотя все они имели соответствующую спецификацию. У 24 из 40 препаратов был превышен рекомендованный компанией Abbott 3% лимит посторонних примесей.
В 10 раз увеличено количество твердых частиц в 4 воспроизведенных препаратах цефотаксима по сравнению с оригинальным лекарственным средством (Клафоран, Hoechst). Указанные частицы, содержащиеся в генериках, могут нарушать микроциркуляцию в ишемизированных тканях и способствовать развитию респираторного дистресс-синдрома и полиорганной недостаточности у тяжелых пациентов .
В литературе представлены результаты сравнения брендового и генерического клозапина (Клозарил, Novartis Pharmaceuticals и клозапин, Zenith Goldline Pharmeceuticals). В ходе исследования было установлено, что расхождение между указанными психотропными препаратами по фармакокинетическим параметрам наблюдается у 40% пациентов, страдающих шизофренией .
Выявлены существенные различия в биоэквивалентности между брендовыми препаратами амитриптилина гидрохлорида, нортриптилина гидрохлорида, дезипрамина, тримипрамина малеата и их генериками .
Проведено более 100 исследований, посвященных биоэквивалентности различных генериков фенитоина, препаратов вальпроевой кислоты, в которых обнаружены существенные расхождения по фармакокинетическим параметрам оригинальных и воспроизведенных лекарственных средств .
Говоря о терапевтической эквивалентности, следует упомянуть исследование R. Mofsen и соавт., в котором описываются 7 случаев неудачной замены у пациентов со стабильным психическим состоянием, находившихся в психоневрологическом интернате, брендового клозапина на его генерик. Подчеркивается, что указанное изменение терапии было неожиданно произведено аптекой и ни врачи, ни медицинский персонал учреждения не знали об этом. Для них оказалось полной неожиданностью возобновление у пациентов психотических расстройств, выраженность которых в 5 из 7 случаев потребовала неотложных мер по переводу больных в психиатрический стационар. Сообщается об аналогичном случае при переводе с брендового пароксетина (паксила) на его генерик .
В проведенном недавно опросе неврологов (301 опрошенный), работающих в США, установлено, что при переводе с брендовых противоэпилептических препаратов на генерические 204 (67,8%) из них наблюдали возобновление судорожных припадков, 168 (55,8%) отмечали усиление побочных эффектов .
Описаны 11 наблюдений, в которых после замены брендового ламотриджина на его генерики контроль над эпилептическими припадками был утрачен.
В результате этих исследований в ряде стран, в том числе в Норвегии, приняты решения, ограничивающие перевод пациентов с брендовых противоэпилептических на генерические препараты, а в Германии эта процедура вообще не рекомендуется .
В ряде контролируемых исследований показано, что при переходе с брендового карбамазепина на его генерик отмечается внезапное возобновление судорожных припадков .
В другой работе, опубликованной в Американском кардиологическом журнале в мае 2000 г., приводится мнение 64 экспертов-электрофизиологов, членов Североамериканского общества стимуляции электрофизиологии, которые сообщают о 32 случаях развития рецидива аритмии (фибрилляции желудочков, желудочковой тахикардии, фибрилляции предсердий и предсердной тахикардии) при замене брендового антиаритмического препарата амиодарона (Кордарон, Sanofi-Synthelabo) на его генерики.
Следует отметить, что имеются также и публикации о терапевтической эквивалентности оригинальных и генерических лекарственных средств. В одном из рандомизированных двойных слепых исследований изучали две параллельные группы амбулаторных пациентов с хронической шизофренией, получавших брендовый флуфеназина деканоат. Первая группа была переведена на его генерик, вторая – оставлена на оригинальном препарате. Через 12 недель в обеих группах отсутствовала какая-либо значимая динамика в состоянии, определяемая по специальной шкале позитивного и негативного синдрома .
Говоря о хронических заболеваниях, необходимо заметить, что многие из них имеют тенденцию к рецидивированию. Ввиду этого современные рекомендации предусматривают наряду с купирующей осуществлять и длительную поддерживающую терапию. На практике нередко наблюдается ситуация, когда купирующая терапия, осуществляемая чаще всего в стационаре, проводится оригинальным лекарственным средством. В дальнейшем после выписки пациента этот препарат в силу «экономических» соображений нередко заменяется на его генерик. В свете представленных выше данных очевидно, что рассматриваемая замена возможна лишь при наличии уверенности в фармацевтической, фармакокинетической и терапевтической эквивалентности оригинального и воспроизведенного лекарственных средств.
Имеются сообщения о том, что появление на фармацевтическом рынке воспроизведенных лекарственных средств не всегда ведет к снижению прямых затрат здравоохранения. В недавно проведенном канадском исследовании проанализировано, что 11% разница в количестве рецидивов, наблюдающаяся при лечении больных генерическим и оригинальным клозапином, сводит на нет преимущество генерика в цене. Аналогичные данные были получены и для противоэпилептических препаратов .
Приведенные данные, как и многие другие, по мнению главного клинического фармаколога МЗ РФ профессора Ю.Б. Белоусова, развеивают миф о дешевизне генериков, так как затраты при их применении гораздо выше, чем при использовании оригинальных препаратов. Вопреки расхожим утверждениям о том, что воспроизведенные лекарственные средства снижают прямые затраты на лечение, способствуют развитию конкурентной борьбы и снижению цен на препараты-бренды и даже являются одним из способов внедрения экономически эффективных медицинских технологий в клиническую практику, некоторые современные исследования свидетельствуют об обратном.
Ученый полагает, что переход с недорогих генериков на оригинальные препараты выгоден как для пациентов, так и для общества в целом. Он считает, что недопустимо переносить данные по эффективности и безопасности, полученные на оригинальных препаратах, на их копии. Лишь наличие полной информации о соблюдении требований GMP при производстве генерика, его фармакокинетическая и терапевтическая эквивалентность при сравнении с оригинальным препаратом делают обоснованным поиск фармакоэкономических преимуществ генерика. В противном случае формально выгодные ценовые показатели могут обернуться огромными дополнительными расходами, например на лечение нежелательных побочных явлений. По мнению Ю.Б. Белоусова, практика, сложившаяся в РФ, разрешающая медицинское применение генерика на основании данных только его биоэквивалентности, неверна. Для определения терапевтической эквивалентности необходимо проведение как ограниченных, так и крупных клинических исследований эффективности генерика при конкретном заболевании, изучение сравнительной эффективности оригинального и воспроизведенного препаратов с использованием четких конечных критериев. Терапевтическая эквивалентность означает и организацию исследований профиля безопасности генериков с интенсивным мониторингом в течение 5 лет после регистрации нежелательных эффектов.
Очевидно, что оригинальные препараты всегда будут противопоставляться генерическим, но их конкуренция на фармацевтическом рынке должна базироваться на строгом соблюдении требований к качеству производства как оригинальных, так и воспроизведенных лекарственных средств, на результатах анализов биоэквивалентности, а также данных клинических исследований. Поэтому широкое использование генерических препаратов в клинической практике должно основываться на доступных практикующим врачам ясных указаниях на их фармацевтическую, фармакокинетическую и в первую очередь терапевтическую эквивалентность оригинальным препаратам.
Список литературы находится в редакции
Генерический лекарственный препарат должен отвечать следующим требованиям:
- · содержать то же активное вещество в той же дозе и лекарственной форме, что и оригинальный лекарственный препарат;
- · быть идентичным оригинальному лекарственному препарату по силе действия;
- · иметь те же показания к применению, что оригинальный лекарственный препарат;
- · быть биоэквивалентным оригинальному препарату (т.е. после перорального приема то же количество лекарственного средства должно иметь такую же концентрацию в крови, что и оригинальный лекарственный препарат).
Если препараты неэквивалентны в биологическом смысле из-за различной технологии изготовления и / или наличия неодинаковых вспомогательных ингредиентов и наполнителей, то возможно различие (неэквивалентность) их лечебного эффекта. Поэтому при сравнении препаратов различных фирм основными в фармакологической характеристике являются понятия биоэквивалентности, фармацевтической эквивалентности и альтернативности, терапевтической эквивалентности.
Фармацевтически эквивалентные лекарственные препараты - препараты в одинаковой лекарственной форме, которые содержат одни и те же активные субстанции в одинаковом количестве, отвечающие требованиям одних и тех же или сходных стандартов. В США фармацевтически эквивалентными считаются лекарственные препараты, которые содержат одинаковые активные ингредиенты в одинаковой лекарственной форме, предназначены для одного способа введения и являются идентичными по силе действия или концентрации активных веществ.
Фармацевтически альтернативные лекарственные препараты - препараты, которые содержат одинаковое лекарственное вещество, но отличаются по химической форме этого вещества (являются разными солями, эфирами или комплексами этих веществ), лекарственной формой или силой действия.
Биоэквивалентные лекарственные препараты - препараты, которые дают одинаковую концентрацию действующих веществ в крови и тканях организма при введении препаратов в равной дозе одним и тем же путем.
В ЕС два лекарственных препарата считаются биоэквивалентными, если они фармацевтически эквивалентны или альтернативны и если их биодоступность (скорость и степень всасывания) после введения в одинаковой молярной дозе сходна в такой степени, что их эффективность и безопасность в основном одинаковы.
В США биоэквивалентные лекарственные препараты определяются как фармацевтически эквивалентные или альтернативные препараты, которые имеют сравнимую биодоступность при исследовании в сходных экспериментальных условиях.
Биоэквивалентность означает, что биоэквивалентные оригиналу препараты-генерики обеспечивают одинаковый фармакодинамический эффект, одинаковую эффективность и безопасность лекарственной терапии.
Исследование биоэквивалентности необходимо для подтверждения качества препаратов-генериков и их соответствия оригинальному препарату.
Подразумевается, что биоэквивалентные препараты являются терапевтически эквивалентными.
Терапевтически эквивалентные лекарственные препараты - препараты, которые содержат одинаковую активную субстанцию или лекарственное вещество и, по результатам клинических исследований, обладают одинаковой эффективностью и безопасностью. При определении терапевтической эквивалентности изучаемый препарат сравнивается с препаратом, чья эффективность и безопасность уже установлены и общепризнаны.
Терапевтически эквивалентными лекарственные препараты могут считаться только в том случае, если они фармацевтически эквивалентны. В таком случае можно ожидать, что они будут иметь одинаковый клинический эффект и одинаковую безопасность при их назначении пациентам.
С понятием биоэквивалентности тесно связано понятие биодоступности.
Биодоступность - часть препарата, попадающая в системный кровоток при внесосудистом пути введения.
При внутрисосудистом введении лекарственное вещество полностью попадает в кровеносное русло и его биодоступность равна 100%. При других путях введения (даже при внутримышечном и подкожном) биодоступность почти никогда не достигает 100%, так как лекарство должно пройти через ряд биологических мембран клеток (слизистой оболочки желудка, печени, мышц и т.д.), и только часть его попадает в системный кровоток. Действие препарата во многом зависит от того, насколько велика эта часть.
Факторы, влияющие на биодоступность:
- · путь введения препарата;
- · индивидуальные особенности организма больного;
- · состояние желудочно-кишечного тракта, сердечно-сосудистой системы, печени, почек;
- · биофармацевтические факторы (лекарственная форма, состав вспомогательных веществ, особенности технологии производства препарата).
Препараты, содержащие одни и те же лекарственные вещества, но выпускаемые различными фармацевтическими фирмами, могут существенно различаться по биодоступности. Различия в биодоступности приводят к различиям в терапевтической эффективности и различной частоте и выраженности побочных эффектов.